CircRNA:
Figure
source:
https://en.wikipedia.org/wiki/Circular_RNA
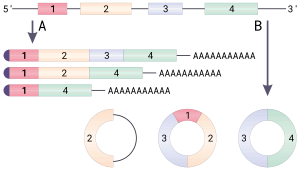
Circular RNAs (circRNAs) are a class
of abundant and conserved RNAs in animals and plants
[1-3]
1. Chen, L. L (2016). The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol 17, 205-211.
[doi:10.1038/nrm.2015.32]
2. Barrett, S. P. & Salzman, J (2016). Circular RNAs: analysis, expression and potential functions. Development 143, 1838-1847. [doi:10.1242/dev.128074]
3. Li, X., Yang, L. & Chen, L. L (2018). The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell 71, 428-442. [doi:10.1016/j.molcel.2018.06.034]
. Most circRNAs are produced from
a special type of alternative splicing known as back-splicing, and are predominantly localized
in cytoplasm
[3-5]
3. Li, X., Yang, L. & Chen, L. L (2018). The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell 71, 428-442.
[doi:10.1016/j.molcel.2018.06.034]
4. Jeck, W. R. et al (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141-157. [doi:10.1261/rna.035667.112]
5. Salzman, J., Gawad, C., Wang, P. L., Lacayo, N. & Brown, P. O (2012). Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7, e30733. [doi:10.1371/journal.pone.0030733]
. However, the general function of circRNA in vivo is still an open question.
Several circRNAs have known functions including sequestration of miRNAs
[6,7]
6. Hansen, T. B. et al (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495, 384-388.
[doi:10.1038/nature11993]
7. Memczak, S. et al (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333-338. [doi:10.1038/nature11928]
or RNA binding proteins (RBPs)
(i.e., as competitors of the linear mRNAs), and modulation of transcription
and interference with splicing
. Nevertheless, the function of the remaining circRNAs is
an uncharted territory and has not benefitted as much from the advancement in sequencing
technology until lately. Since in vitro synthesized circRNAs can be translated in
cap-independent fashion
[11]
11. Chen, C. Y. & Sarnow, P (1995). Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268, 415-417.
[doi:10.1126/science.7536344]
and many protein-coding genes in higher eukaryotes can produce
circRNAs through back-splicing of exons, it is highly possible that they function as mRNAs
in vivo to direct protein synthesis. Recent studies indicated that some cytoplasmic
circRNAs can be effectively translated into detectable peptides, and many short sequences,
including m6A modification sites have been suggested to function as IRES-like elements to
drive circRNA translation
. Using various direct and indirect evidences that support
circRNA translation, we conducted an integrative analysis to predict the potential of all
circRNAs in coding for functional peptides. The result of such prediction and the supporting
evidences were summarized in the TransCirc database.
Motivation of TransCirc database:
Now as translation of circRNAs is starting to grow up a new field, more and more researchers would
benefit from a database collecting circRNAs that can translate. We noticed that a translatable
circRNA database is still lacking, and hope to fill the gap to make circRNA translation studies more
convenient.
Evidences of circRNA translation:
1. Ribosome/polysome profiling
The translation of mRNAs is carried out by ribosome, which can form polysomes in
actively translated mRNAs. Therefore the association with ribosomes/polysomes can serve as a strong
predictor of the potential for translatable circRNA. We used published ribosomal foot printing data
and polysomal profile
to score the association of circRNAs with
ribosomes, which can serve as a strong predictor for translation of circRNAs.
2. Translation initiation site (TIS)
A global mapping of TIS codons at nearly single-nucleotide resolution has been
achieved by GTI-seq
[18]
18. Sooncheol Lee, Botao Liu, et al.
Proceedings of the National Academy of Sciences of the United States of America 2012, 109:E2424-32.
[doi:10.1073/pnas.1207846109]
, revealing an
unambiguous set of several thousands TIS codons across the entire human transcriptome. We used the
data from TISdb based on GTI-seq as an indirect evidence supporting the translation of circRNAs,
which also associated with potential ORFs.
3. IRES sequence
Since circRNAs are covalently closed molecules without free ends, the translation of
circRNAs must use an unconventional initiation mechanism known as cap-independent translation
initiation. Such initiation pathway has to be driven by IRESs (internal ribosomal entry sites),
which are typically short RNA fragments with specialized secondary structure. Although most
well-studied IRESs were found in various virus RNAs, there are several cases where the endogenous
genes contain IRES to drive translation in a cap-independent fashion. Recently, we and other group
conducted a systematic screen for IRES elements in human genome or from random sequences
, and
thus we used all the available IRES information as evidence to support circRNA translation.
4. m6A sites
The N-6-methyladenosine (m6A) is the most common modification of RNAs, and have been
found in many types of non-coding and coding RNAs. We have recently found that circRNAs undergo
extensive modification m6A, which can drive circRNA translation through recruiting reader protein
YTHDF3 that interacts with translation initiation factors (most notably eIF4G2). Therefore we used
published m6A modification data from REPIC database
(identified by three different tools), and mapped them back to circRNA
sequences. The existence of experimentally validated m6A sites in circRNA can also serve as a
predictor for translatable circRNA
, which
are integrated in this database.
5. ORF length
The length of potential open reading frame (ORF) is a common predictor for coding RNA
vs. non-coding
RNAs. Usually a long ORF cannot be found in a non-coding RNA, and thus we used the ORF length > 20aa
as a minimal requirement for circRNA encoded peptide. It should be noticed that ORF length is a
relatively weak predictor, as many small peptide were recently found to be coded by “non-coding”
RNAs in human transcriptome, whereas circRNAs with a long ORF will have better chance to be a coding
mRNA.
6. sequence composition
The amino acid (aa) sequences of all natural proteins only occupy a very small
fraction of the possible sequence space, mostly because only a sub-fraction of sequences can form
stable proteins. Therefore the protein with “unnatural” sequence tend to be degraded rapidly, and
the sequence similarity to all natural proteins can serve as a strong predictor to identify
authentic proteins in random strings of amino acid sequences. Therefore we used machine learning
approach to predict how likely a given sequence is with natural proteins, and applied this
prediction to score for how likely a given ORF encoded by circRNAs can serve as a template for
functional protein.
7. Proteomics evidence by Mass spectrometry
Mass spectrometry is an important method to accurate identify and characterize
proteins. Several large scale mass-spectrometry experiments have been conducted to study human
proteome {}, however only about ~50% of MS spectra can be reliably assigned to known peptides coded
by human mRNAs even considering the post-translational modification of protein. This result suggests
that there are a large fraction of “hidden proteome” encoded by non-canonical mRNA, some of which
could be coded by circRNAs. We have defined a new set of rules and rigorous filters to search
circRNA-encoded peptide from MS dataset, and use the search results as a strong evidence for circRNA
translation. We included all the raw MS spectra supporting the circRNA encoded peptides that cross
the back-splice junctions.
Data Content of TransCirc database:
Context information: taxonomy
host gene information: genome build, position, official name, etc.
circRNA information: locations, exons and sequences
ORF information: starts and ends, sequences
Protein/peptide product information: sequences, prediction of being protein-alike and mass
spectrometry
Data sources:
circRNA:
Its taxonomy, host gene and location information were collected from circAtlas.
Ribosomal and polysome binding evidence:
ribosomal foot printing data
polysomal profile
Translation initiation site:
TISdb based on GTI-seq
IRES sequences:
IRES elements in human genome or from random sequences
m6A sites:
Usage of TransCirc:
1. Simple Search
Search by Ensembl gene: e.g. IQCJ-SCHIP1 | ENSG00000283154.1
Search by Transcirc ID: e.g. TC-hsa-IQCJ-SCHIP1_0001
Search by other circRNA ID: e.g. hsa_circ_03218 | hsa-circRNA5793
Search by genomic position: e.g. chr3:159764000-159766000
2. Sequence based search
3. Browse and filter
The browse and filter page lists all potentially translatable circRNAs based on the
user's query. Users can use the evidence filtering and sorting tools at the top of the page to find
circRNAs of interest.
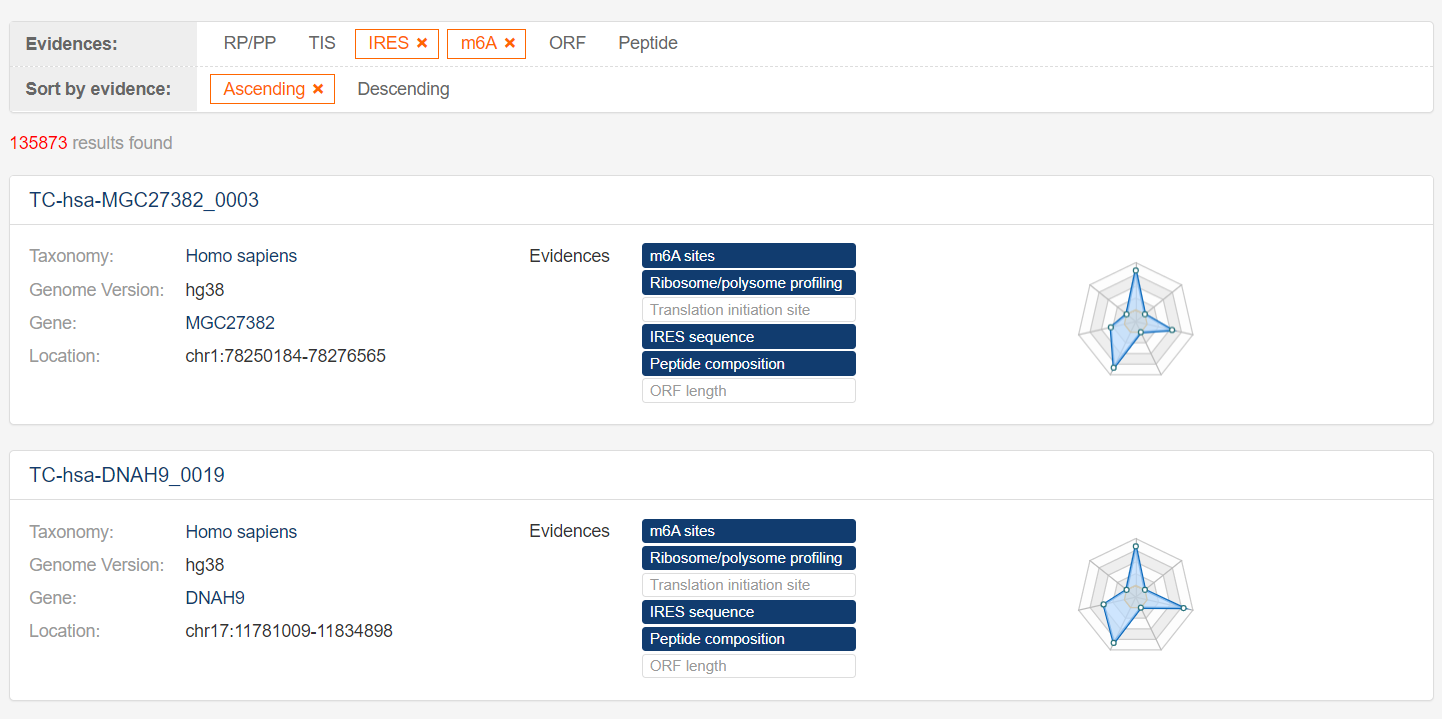
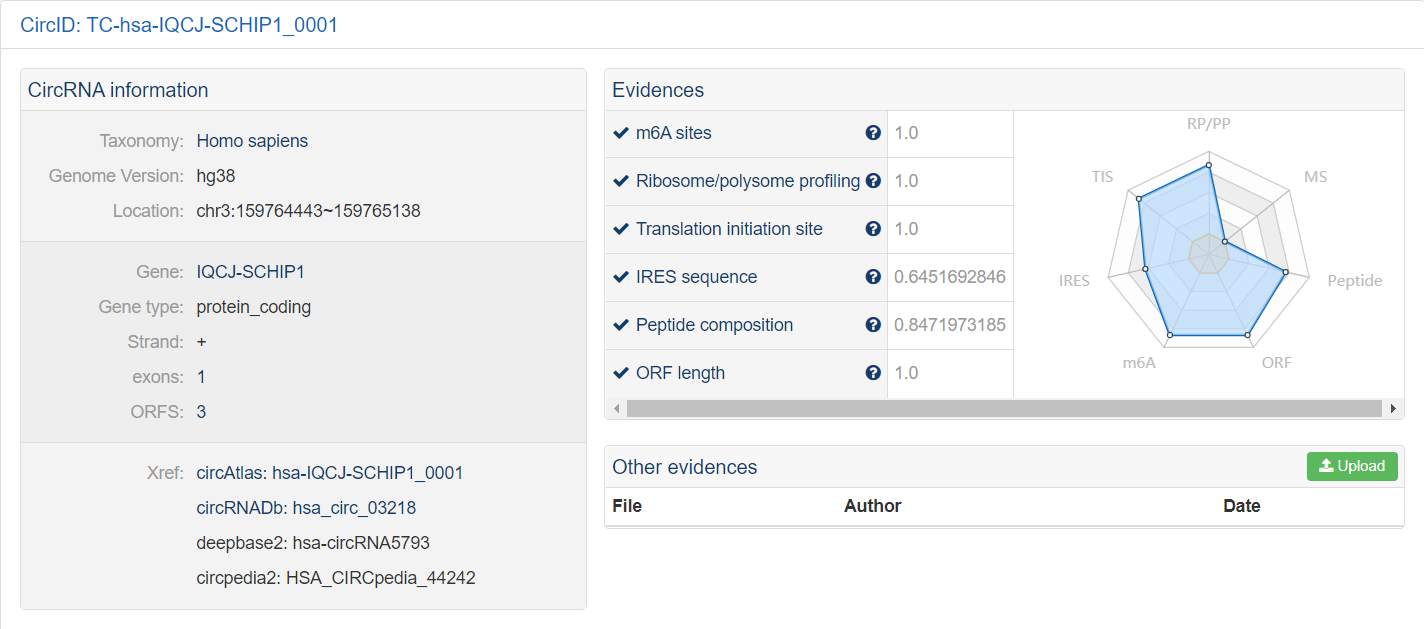
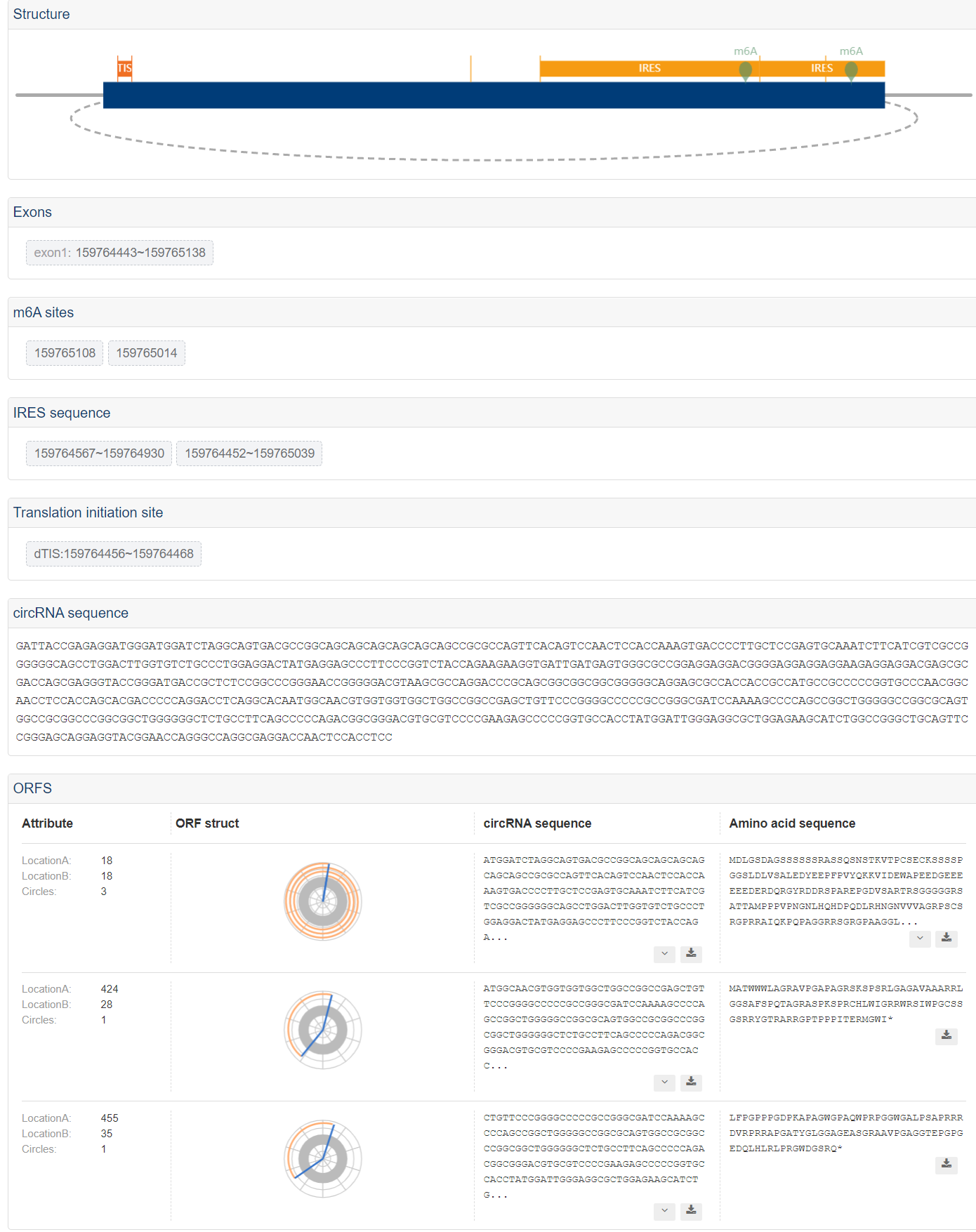
4. Detailed View
View circRNA detailed information, evidences, exon compositions, sequences, ORF locations, predicted
peptides and more. We provided visualization of various aspects of circRNAs and their products.
Please let us know if you wish any further information or representations.
Example of detailed information
Example of evidences for details
5. Downloading