Documentation
Introduction
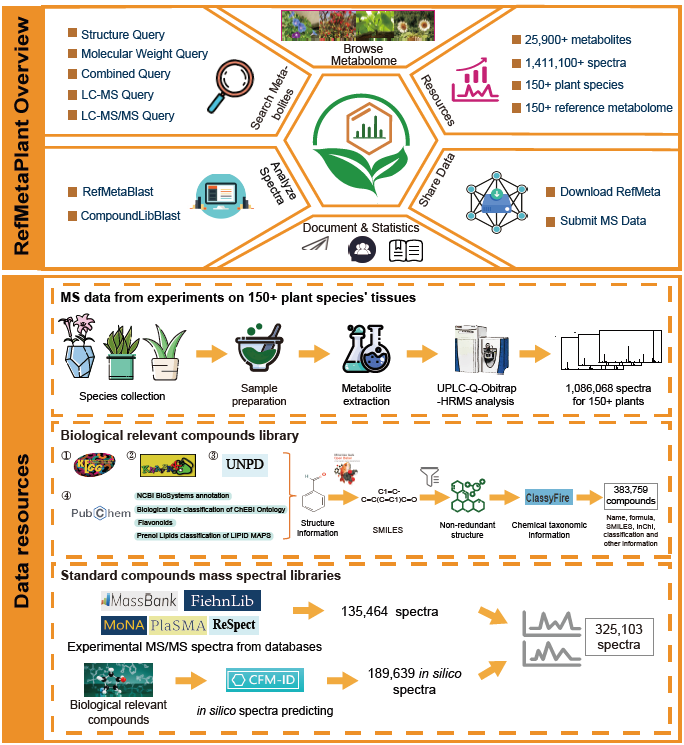
Reference Metabolome Database for Plants (RefMetaPlant) serves as an integrated database and analysis platform dedicated to becoming the centralized resource for plant metabolomic research. It aims to standardize and integrate the reference metabolome data, providing a comprehensive platform for researchers in plant metabolomics, genetics, and related fields. Currently, RefMetaPlant 1.0 is released to provided:
1) 1,086,000+ experimental mass spectra we obtained using UPLC coupled with Quadrupole-Orbitrap High Resolution Mass Spectrometer (UPLC-Q-Orbitrap-HRMS) on samples of 150+ plant species from Bryophyta, Lycopodiopsida, Pteridophyta, Gymnospermae, and Angiospermae;
2) The reference metabolome for 153 plant species across the five major phyla of green plants;
3) 325,100+ standard compounds mass spectral data in a library, which include data of 135,464 experimental reference mass spectral from public databases like MassBank, MoNA, Respect, FiehnLib, RIKEN PlaSMA, and data of 189,639 in silico mass spectra;
4) A set of related query and analytical tools like ‘LC-MS/MS Query’, 'RefMetaBlast' and 'CompoundLibBlast' for plants metabolome search and profiling, and metabolite identification.
RefMetaPlant provides a powerful platform to support plant genome-scale metabolomics analysis, and promote knowledge/data sharing and collaborations of metabolomic research.
 Overview of the RefMetaPlant
Overview of the RefMetaPlant
Data Collection
1. Metabolites
25,912 metabolites from 153 different plant species, which were made up of Lipids, Terpenoids, Carboxylic Acids, Amino Acids, Peptides, Flavonoids, etc.
2. Spectra
(1) Experimental spectral library (1,221,532 spectra)
i) public experimental spectrum library: 135,464 experimental spectra collected from the
public records of MassBank, Respect, Fiehn HILIC, Vaniya and RIKEN PlaSMA.
ii) species-specific experimental spectrum library: 1,086,068 species-specific experimental
spectra of 153 different plant species.
(2) In silico spectral library
The structural data of compounds were collected from four biologically relevant structure
databases, including KEGG, KNApSAcK, PubChem and UNPD. All these structural data were used
to generate in silico mass spectra by CFM-ID software, and corresponding in silico mass
spectra were stored in our in silico spectral library for metabolite annotation.
Data Processing
Plant Metabolome data processing pipelines include peak detection, alignment, annotation and profiling were carried out using the non-targeted MS-analysis protocol with UPLC-Q-Orbitrap mass spectrometer and an integrated bioinformatics pipeline.
1. peak detection and alignment
The raw Metabolome data were processed with Compound Discoverer software (v3.2, Thermo-Fisher Scientific) using its automatic workflow, including peak detection and alignment (Li et al. 2022). The peak detecting parameters were as follows: min peak intensity, 10E6; S/N threshold, 5. The retention time aligning parameters were as follows: mass tolerance,5 ppm; maximum shift, 0.5 min.
2. Metabolite annotation and metabolic profiling
Metabolite annotation mainly adopted two complementary approaches with experimental/in-silico mass spectra as reference, and the Reference Metabolome for each species was profiled using the integrated bioinformatics pipeline in our previous study (Li et al. 2022).
Quick Search
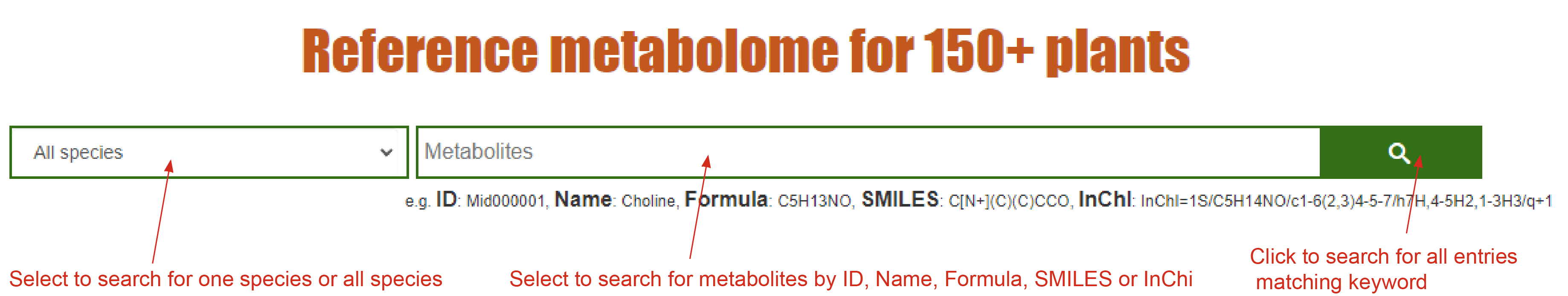
Search the Reference Metabolome for 150+ plants by compound ID, Name, Formula, SMILES, and InChI.
 Quich search
Quich search
You can input key fields of data entries including compound ID, Name, Formula, SMILES or InChI in the search bar for quick search. The sub-query box will provide options to search in each species or all the species in RefMetaPlant. A search result page containing all the matching records will return to the users, and users can click ‘Display full record’ to display the detail information of each compound.
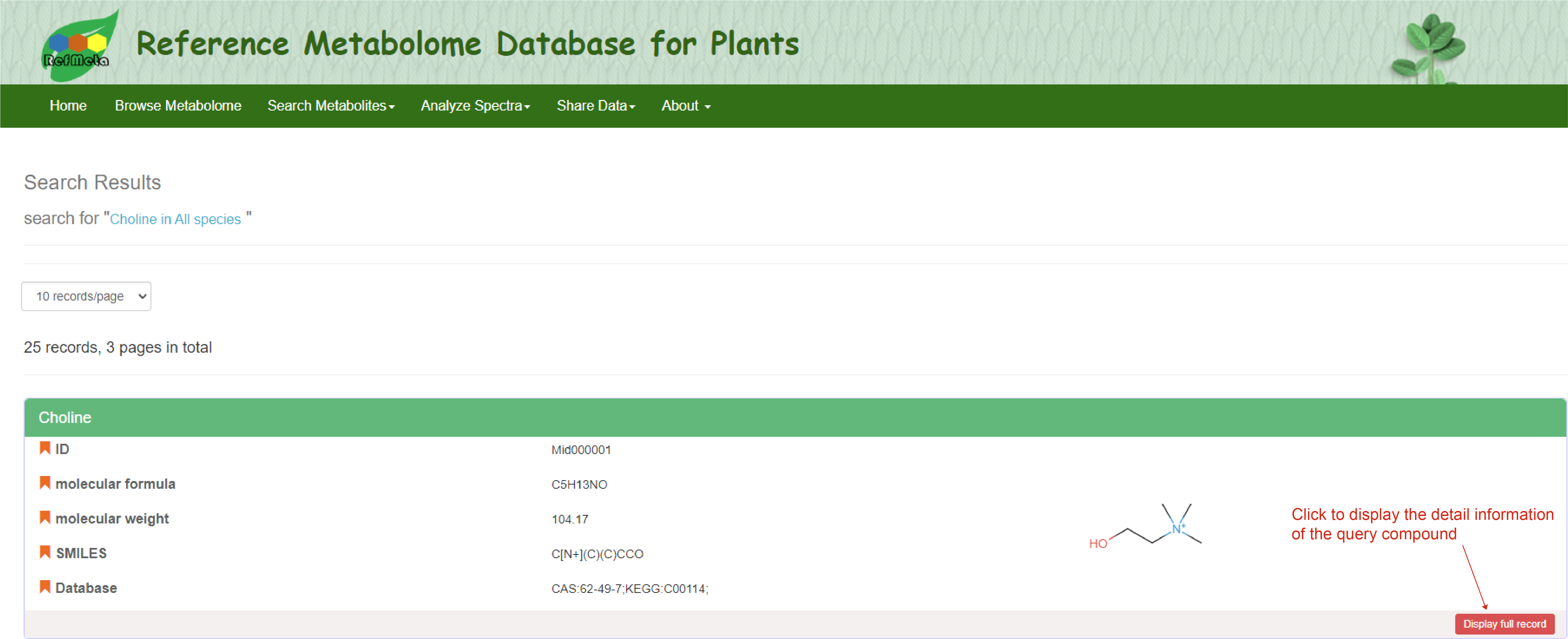 Quick search result
Quick search result
Browse Metabolome
The ‘Browse Metabolome’ module is designed to exhibit the reference metabolome for 153 plant species ranging from Bryophyta to Angiospermae. Users can browse the webservice via clicking the "Browse Metabolome" button on the homepage, and then access the reference metabolome for each plant species. By clicking the picture of each species, users can obtain the reference metabolome of all the metabolites that have been identified for corresponding species. The page displays all the metabolites in the reference metabolome of each species, including the total number. Detailed information of each metabolite can be accessed by clicking the "Display full record". The detail information about materials and analytical conditions can be found in metadata. In the "metadata" page, users can view the information of sample set, sample, sample preparation and analytical method.
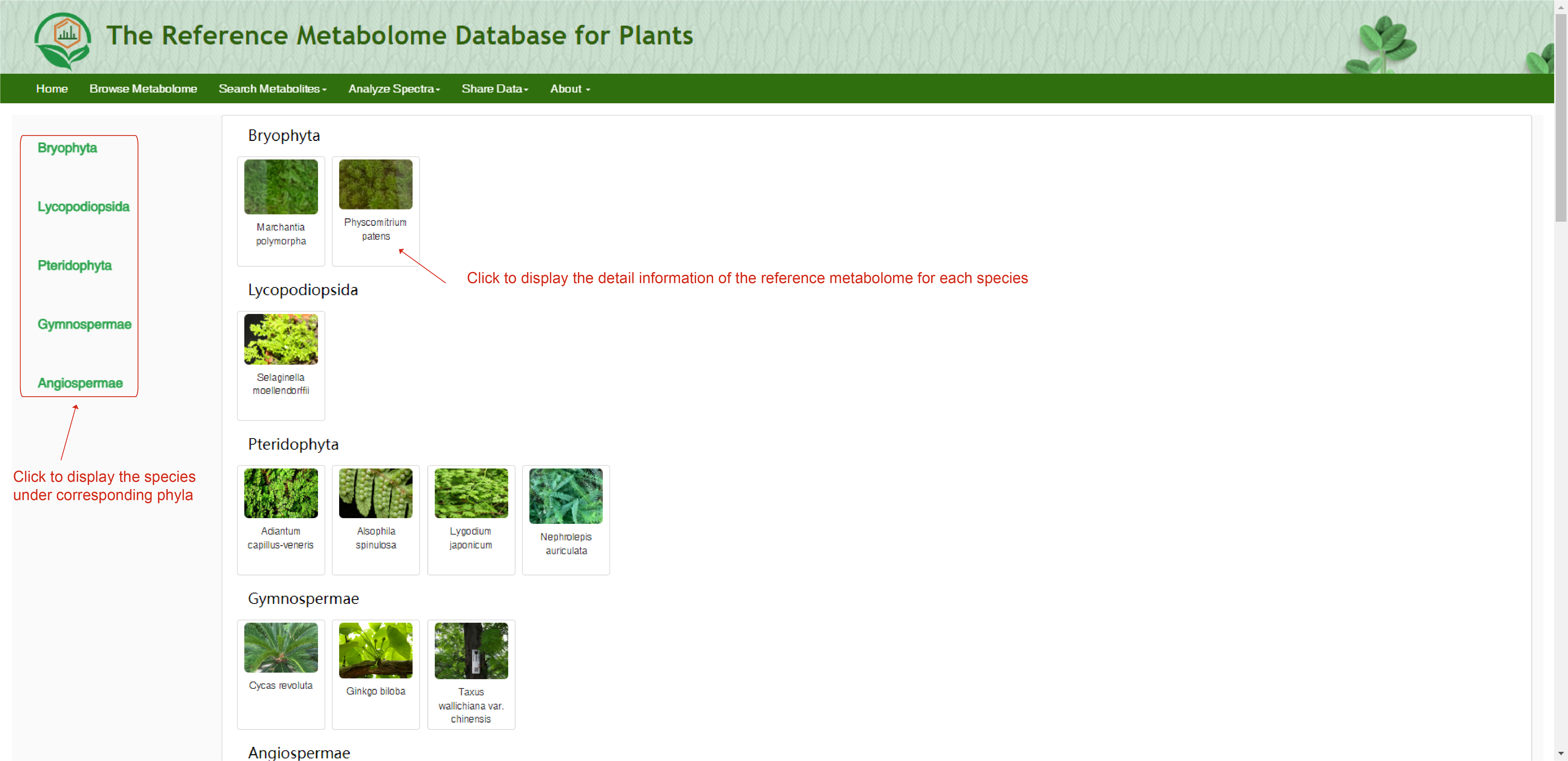 Browse Metabolome page
Browse Metabolome page
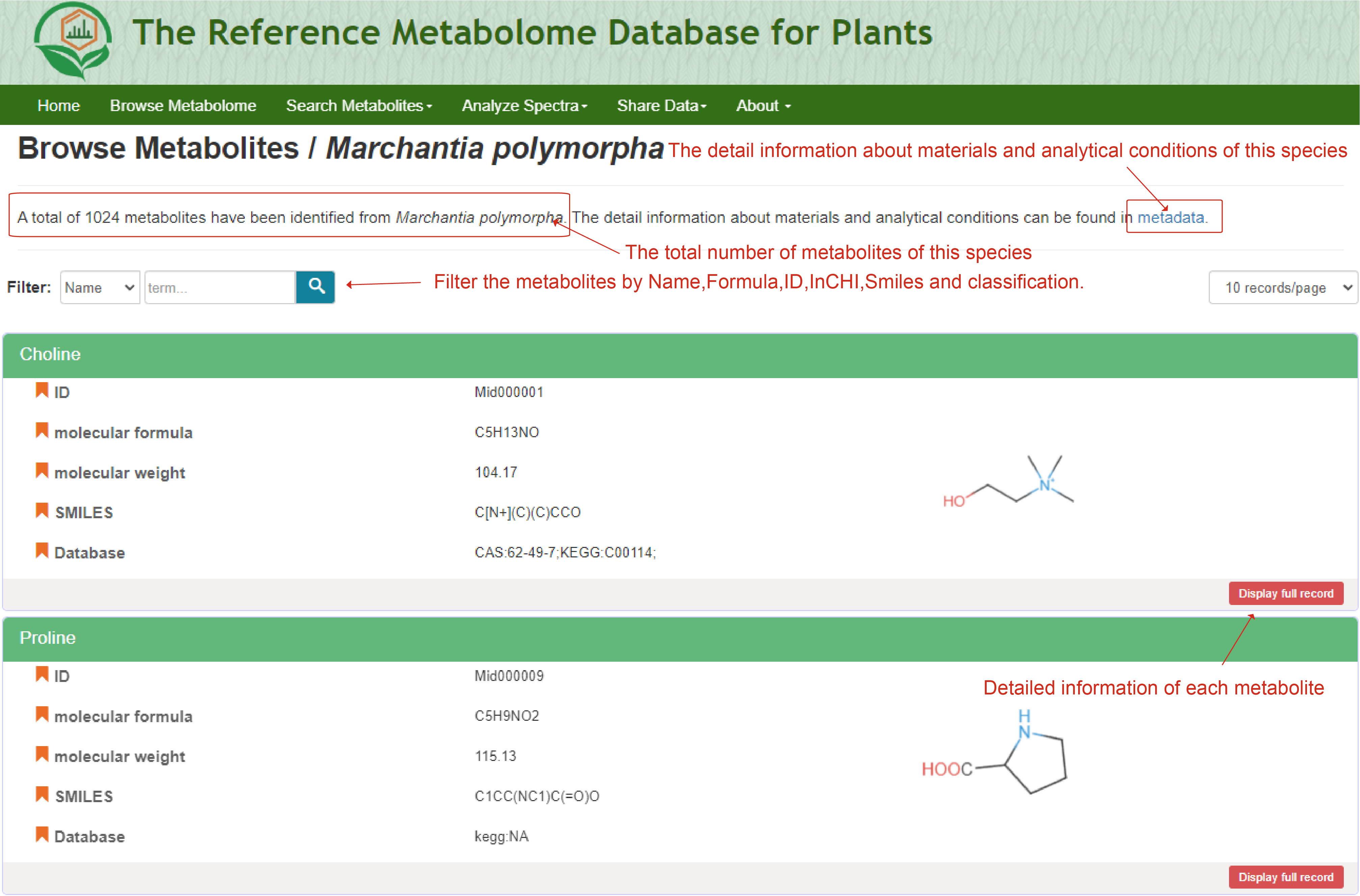 The reference metabolome for interested plant species
The reference metabolome for interested plant species
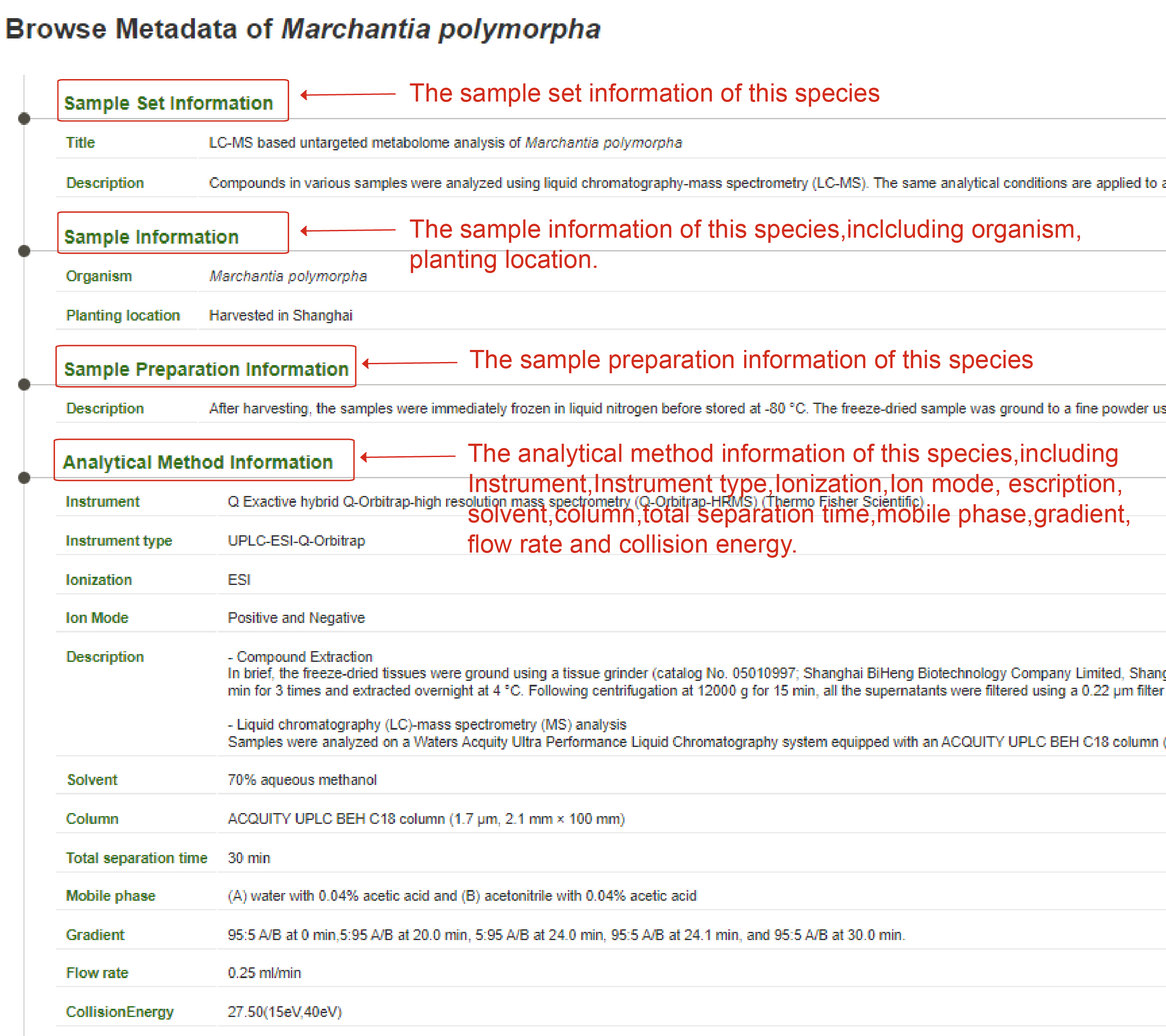 The detail information about materials and analytical conditions for interested plant species
The detail information about materials and analytical conditions for interested plant species
Search Metabolites
Structure query
Users can use the following tool box to sketch molecular structure of a metabolite as a query to search for related metabolites in Reference Metabolome Database. After clicking the "Search" button, a new webpage will display matched metabolites.
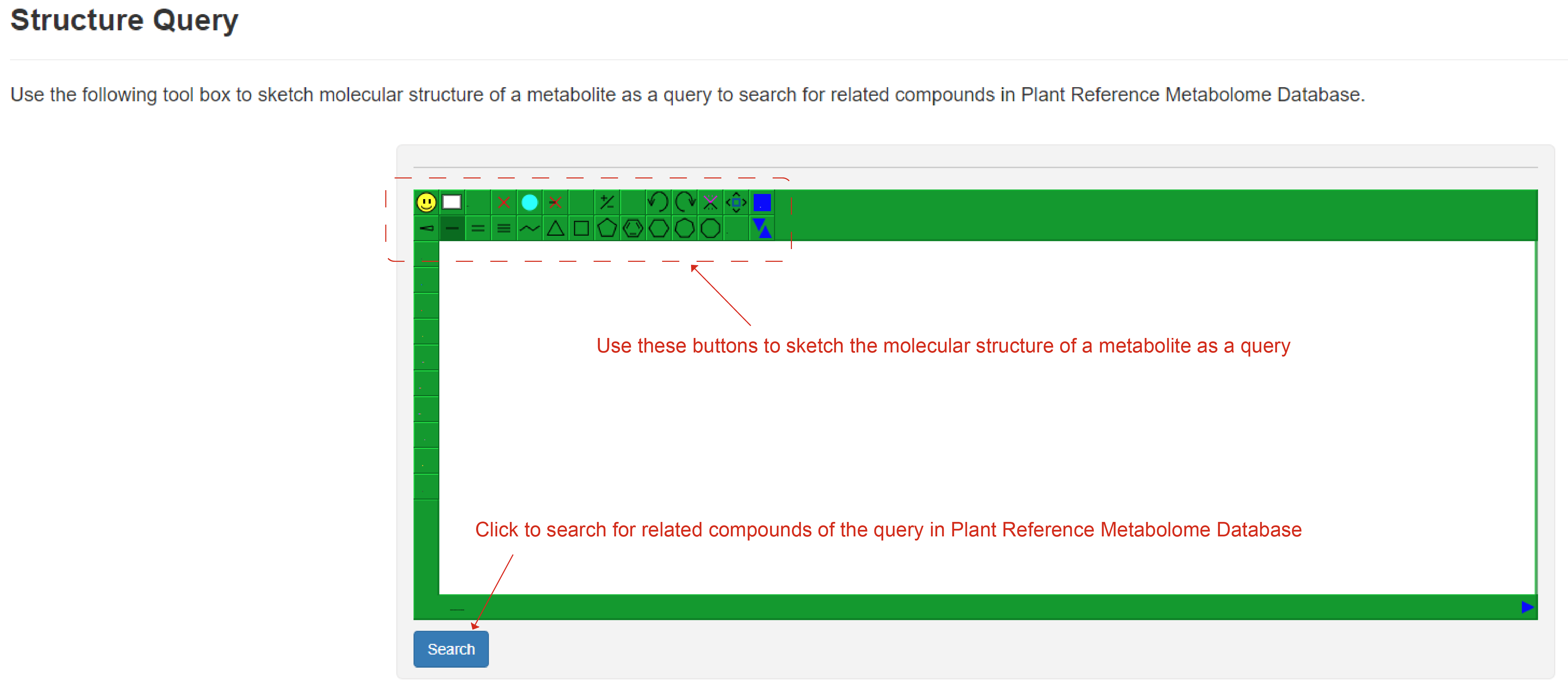 Structure query page
Structure query page
Molecular Weight Query
The molecular weight query allows user to set the range of molecular weight to search for metabolites in Reference Metabolome Database. After clicking the "Search" button, a new webpage will display matched metabolites.
 Molecular weight query page
Molecular weight query page
Combined Query
Combined Query enables users to search for metabolites of their interest by specifying structural properties and molecular descriptors in Reference Metabolome Database. Users can use one or combination of multiple of these options in "Name, Formula, ID, Smiles, InChl, Class and Species" to customize their search. After clicking the "Search" button, a new webpage will display matched metabolites.
 Combined query page
Combined query page
LC-MS Query
LC-MS Query allows users to search against the species-specific experimental spectrum library uses one or multiple m/z values of precursor ions and returns matched metabolites. Clicking the "Load Sample" button will automatically fill in a sample data. In this tool, one or multiple m/z values of precursor ions from sample MS spectra are manually entered in the text box. Then users can set the parameter of m/z tolerance, ion mode, and sample species, before query is executed by clicking the ‘search’ button.
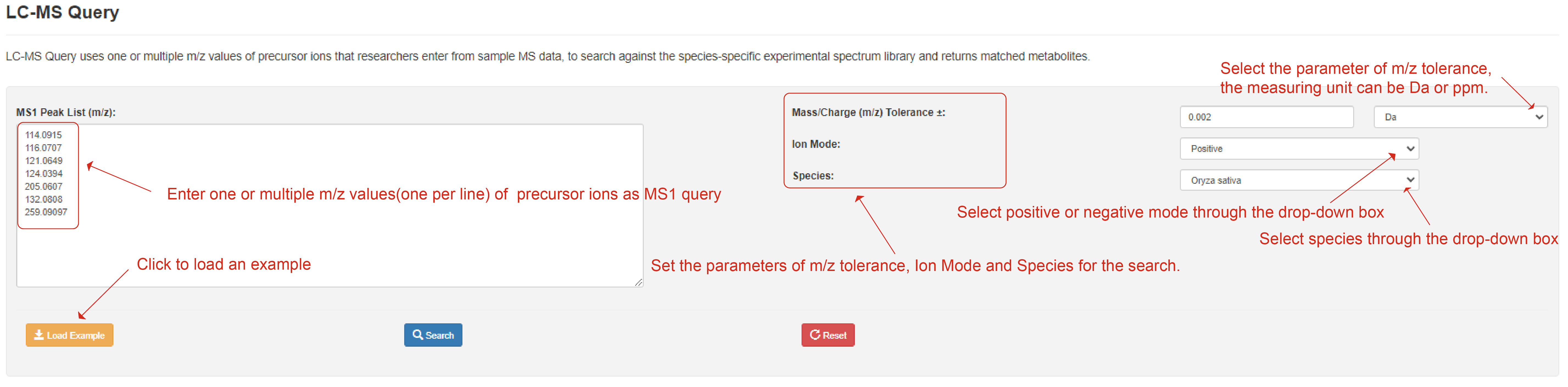 LC-MS Query page
LC-MS Query page
LC-MS/MS Query
LC-MS/MS Query allow users to uses MS1 data (Parent Ion m/z) and MS2 data (Fragment Ions m/z and Intensity values) entered by researchers, to search against either the experimental spectral library (default, both public experimental spectrum library and species-specific experimental spectrum library), or both the experimental spectral library and the in silico spectral library (check select box). Clicking the "Load Sample" button will automatically fill in a sample data. Notably, in the returned results, the matched records are ordered with the similarity scores computed using the INCOS algorithm. It returns all matched reference metabolites for the paired MS1/MS2 data of your interest.
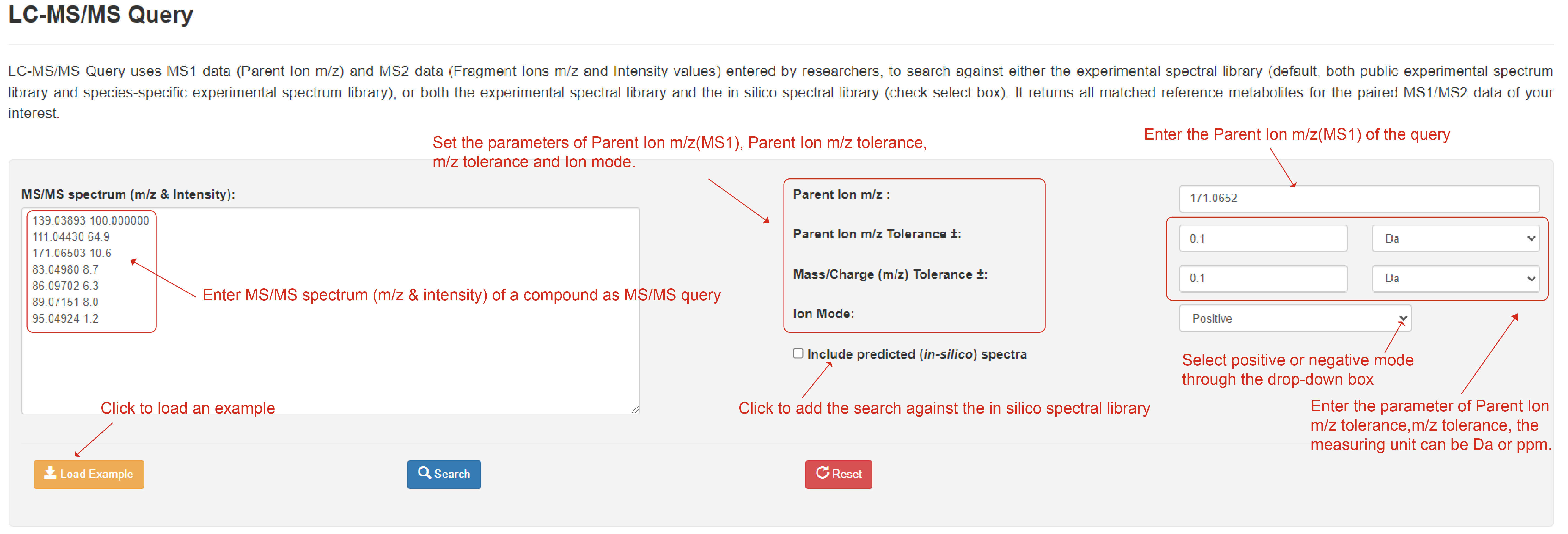 LC-MS/MS Query page
LC-MS/MS Query page
Analyze Spectra
RefMetaBlast
RefMetaBlast allows users to upload sample LC-MS datafiles in standard formats (mzML, mzXML, or mzData), and uses our pipeline to perform metabolite annotation on the samples by comparing with a selected reference metabolome.
The pipeline consists of three steps:
1) detecting peaks in MS1 spectra and extracting MS2 spectra for the detected peaks;
2) annotating peaks by matching their MS1/MS2 patterns to the species-specific experimental spectrum library;
3) reporting the sample metabolic profile by extracting peak intensity values and metabolite identity.
You need to enter your Job title for your analysis, choose a datafile to upload for analyzing and set the parameters for ion mode and the species accurately. Once the data is submitted successfully, you can click the ‘Start analysis’ button to start analysis. When the analysis is complete, a web link to a result page will be on the page. Users can use to retrieve the annotation results that include statistics of annotated peaks, categories of identified metabolites, and downloadable files for all extracted MS/MS spectra, and annotation of non-redundant peaks.
Note: tips for using RefMetaBlast
- 1. Sample LC-MS datafiles must contain high-resolution LC-MS data collected off an instrument with centroid mode.
- 2. Sample LC-MS datafiles from different instruments can be converted to standard formats (mzML, mzXML, or mzData) using third party tools. One commonly used is ProteoWizard.
- 3. One LC-MS datafile can be uploaded at a time; a datafile is limited to 100M in size. An example is found here: .
- 4. RefMetaBlast usually runs from minutes to hours for each LC-MS datafile. After submitting the data analysis, please save the download link provided on the page, the result page can be obtained from this link once analysis is completed.
- 5. Your data are kept confidential with all uploaded data and results being automatically deleted within 72 hours of the completion of the analysis.
- 6. If you are interested to collaborate with us on expanding RefMetaPlant and covering other plants. Please use the "New species submission" tool and feel free to contact us .
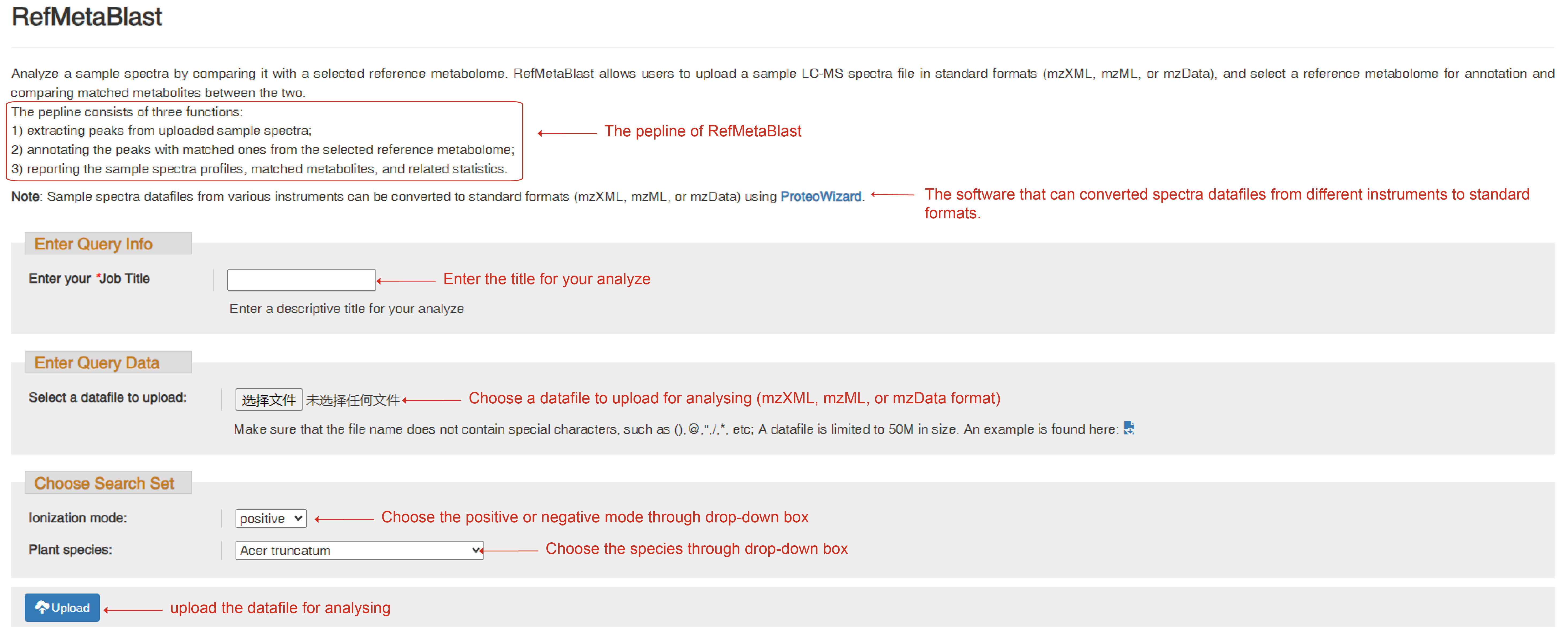 RefMetaBlast page to upload sample LC-MS datafiles
RefMetaBlast page to upload sample LC-MS datafiles
 Analysis interface of RefMetaBlast
Analysis interface of RefMetaBlast
CompoundLibBlast
CompoundLibBlast allows users to upload sample LC-MS datafiles in standard formats (mzML, mzXML, or mzData), and uses our pipeline to perform metabolite annotation on the samples against the compound library. The pipeline consists of three steps:
1) detecting peaks in MS1 spectra and extracting MS2 spectra for the detected peaks;
2) annotating peaks by matching their MS1/MS2 patterns to the experimental spectral library (default, both public experimental spectrum library and species-specific experimental spectrum library), or both the experimental spectral library and the in silico spectral library;
3) reporting the sample metabolic profile by extracting peak intensity values and metabolite identity.
Besides enter your Job title for your analysis, choose a datafile to upload for analyzing and set the parameters for ion mode and the species accurately, you can also click to choose whether annotated with in-silico spectra library.
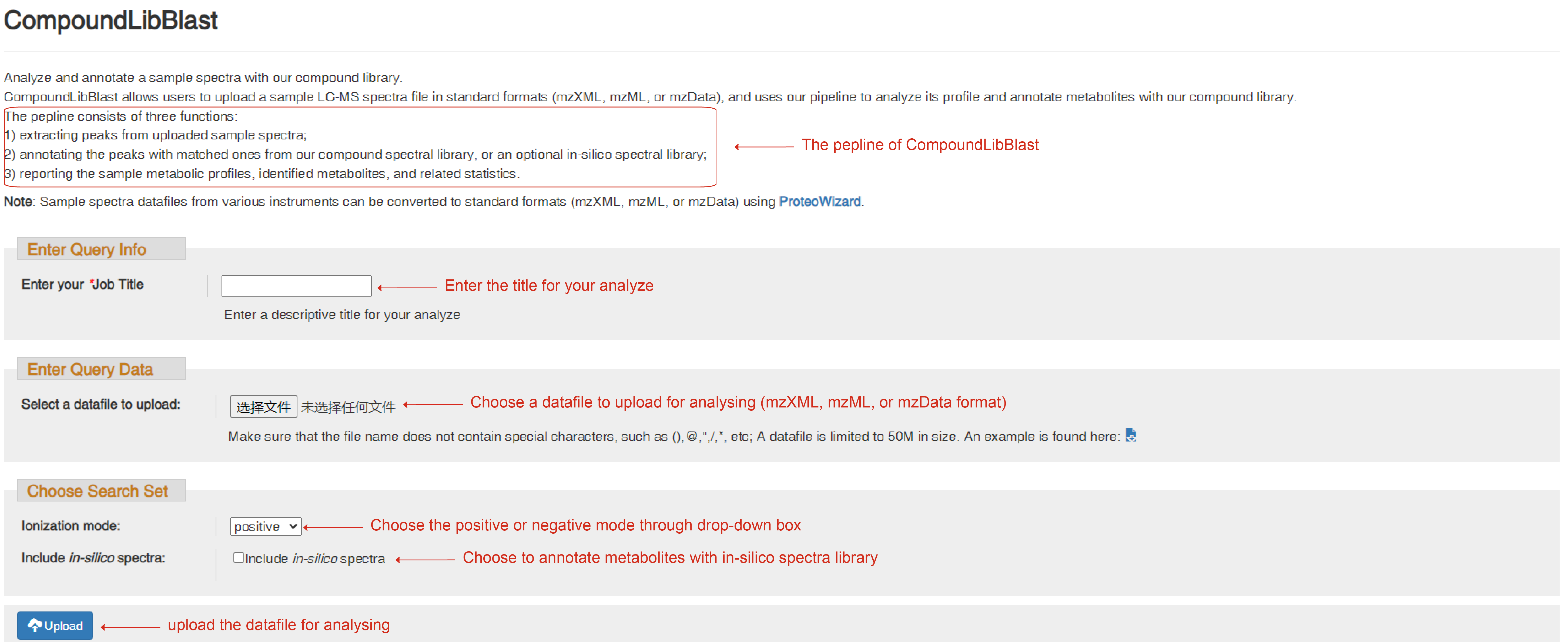 CompoundLibBlast page to upload sample LC-MS datafiles
CompoundLibBlast page to upload sample LC-MS datafiles
 Analysis interface of CompoundLibBlast
Analysis interface of CompoundLibBlast
Share Data
Download RefMeta
The "Download RefMeta" page allows users to download the reference metabolome data for all the 153 plants species. There is a download summary table that includes several data sections of the information of "Species, Genus, Family, Order, Phylum" users can filter depends on their interest. Each reference metabolome can be downloaded in the format of.msr or .mgf, and users can filter the species.
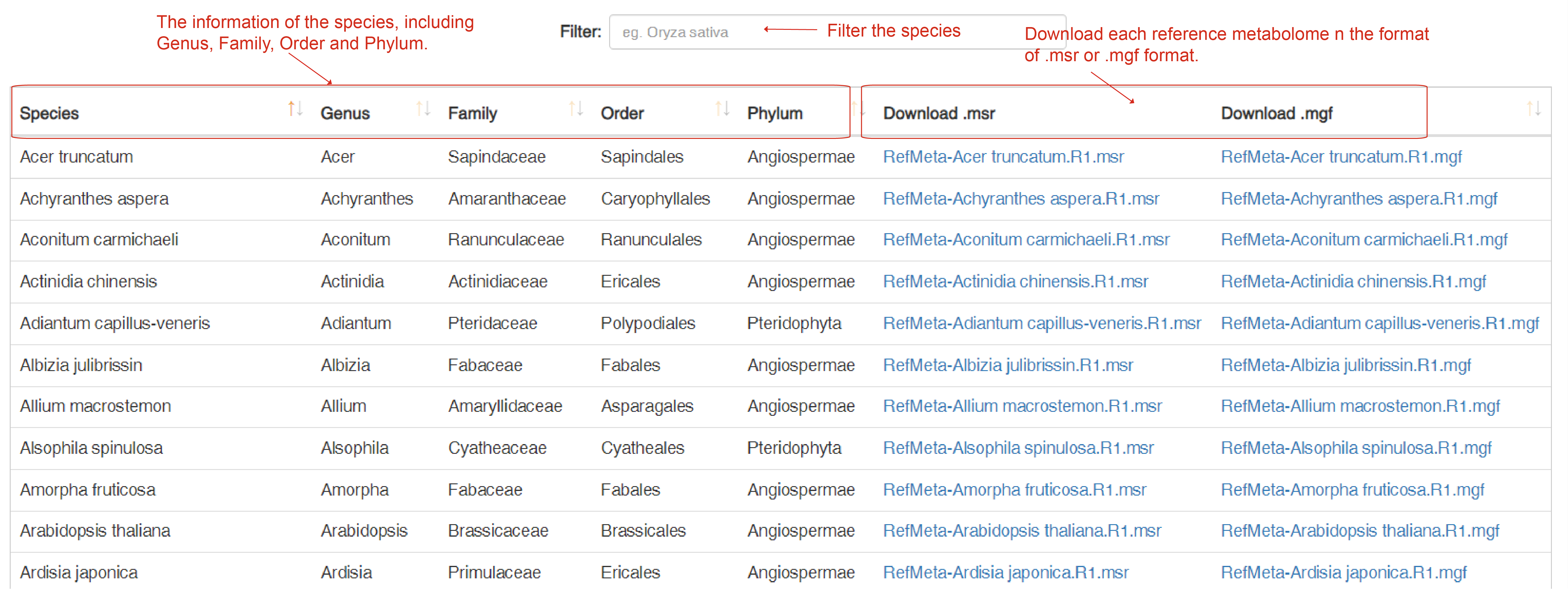 Download RefMeta page
Download RefMeta page
RefMeta-*.R1.msr file
RefMeta-*.R1.msr file has its own format that constitute of the meta data of the species and the MS2 data of the metabolites in the corresponding Reference Metabolome. Meta data contains the information of " DEFINITION, IDENTTFIER, FORMAT , VERSION, KEYWORDS, ORGANISM, CREATION, PUBLICATION, JOURNAL, AUTHORS, And COMMENT". The MS2 data of the metabolites contains the information of "MsLevel, Instrument, InstrumentType, IonMode, CollisionEnergy, PrecursorMz, Annotation, Peak:m/z and Relative Intensity", and each metabolite is divided by "BEGIN" and "END".
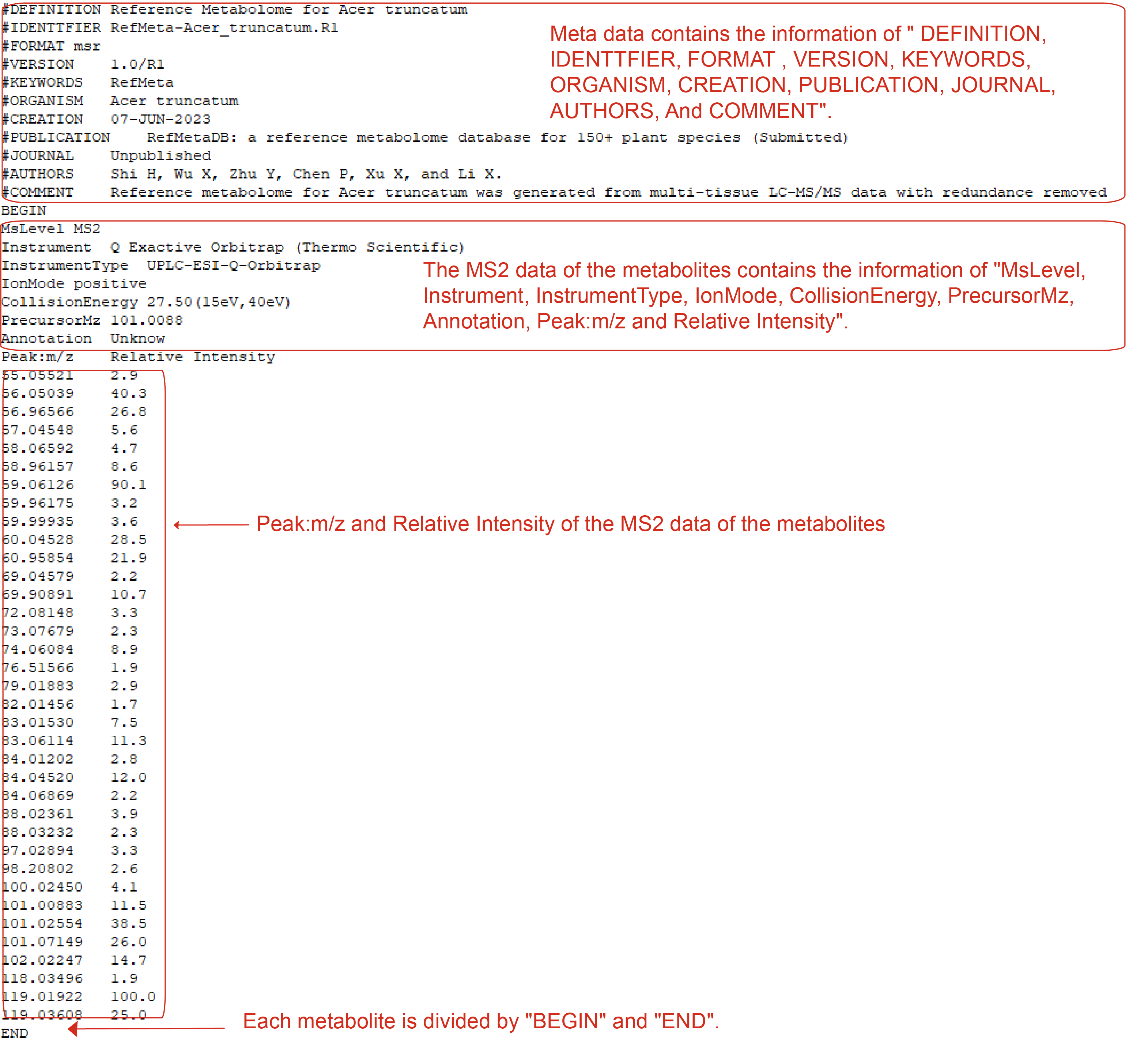 RefMeta-*.R1.msr file example
RefMeta-*.R1.msr file example
Submit MS Data
RefMetaPlant is a public repository for dissemination of plant metabolomics reference data. The "Submit MS Data" page invites users to submit raw metabolomics data for currently existing plants and for others. Note, only mass spectral raw data files are currently supported. Submissions are kept confidential until posted for open access on a date set by submitters.
In order to submit metabolomics data, you need to first contact us to acquire a Project_ID. A filled project-sample-metadata file is needed when you request a Project_ID by email. Once a Project_ID is assigned to you by email, you will be able to submit new data files, or update existing data files under the project you own.
Note, one project can include a number of samples. And each sample can have multiple raw data files because of sample-experiment-polarity-repeat# combinations. Project-sample-experiment relation information is included in the project-sample-metadata file used to accompany data file for submission.
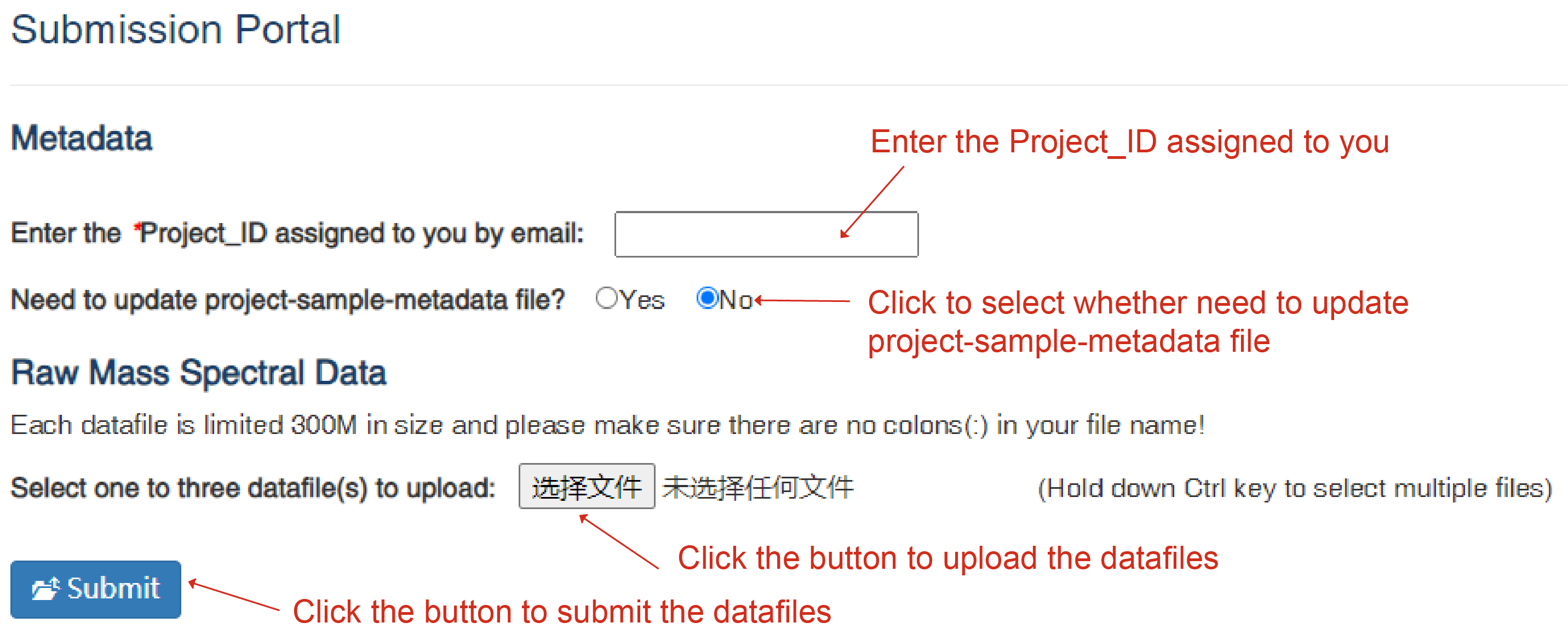 Submit MS Data page
Submit MS Data page
Terms And Abbreviations
Terms and abbreviations commonly used in Reference Metabolome Database for Plants(RefMetaPlant).
- LC-MS
- LC-MS/MS
- Reference Metabolome
- Experimental spectral library
- Public experimental spectrum library
- Species-specific experimental spectrum library
- In silico spectral library
- Metabolite Spectrum Accession Label
- IUPAC
- InChI
- InChI
- SMILES
- PubChem
- CID
- KEGG
liquid chromatography-mass spectrometry.
liquid chromatography tandem-mass spectrometry.
Reference Metabolome of each species. Reference Metabolome of RefMetaPlant contains MS/MS spectra of known metabolites and unknown metabolites from each species.
The experimental spectral library is made up of public experimental spectrum library and the species-specific experimental spectrum library.
Experimental spectra collected from the public records of MassBank, Respect, Fiehn HILIC, Vaniya and RIKEN PlaSMA.
Species-specific experimental spectra of 153 different plant species.
In silico mass spectra of the structural data of compounds collected from four biologically relevant structure databases, including KEGG, KNApSAcK, PubChem and UNPD, generating by CFM-ID software.
Each metabolite spectrum accession was labeled in the format ‘RE0147p′ for example; this denotes the 147th spectrum (0147) derived from the metabolome of rice (RE) extracts obtained in the positive ion mode (p, positive).
International Union of Pure and Applied Chemistry.
IUPAC International Chemical Identifier.
IUPAC International Chemical Identifier.
Simplified Molecular Input Line Entry Specification.
A public database of chemicals and chemical information.
PubChem ID.
Kyoto Encyclopedia of Genes and Genomes.