
CyclicPepedia Knowledge Base
CyclicPepedia (V1.3.1) Tutorial
Table of Contents- Overview
- Browsing interface
- Cyclic peptide details page
- Search tools
- Cyclic peptide tools
- Table 1. List of the chemical and physical properties.
- Table 2. List of the peptide sequence properties.
- Table 3. Sequence formats.
Overview
CyclicPepedia is a pioneering database that encompasses a large amount of known cyclic peptides. This repository houses data on 8744 cyclic peptides, with a wealth of information regarding cyclic peptide sources, categorizations, structural characteristics, pharmacokinetic profiles, physicochemical attributes, patented drug applications, and a collection of relevant publications. Therefore, CyclicPepedia forms a comprehensive knowledge network of cyclic peptides, thereby facilitating advancements in the early stage of cyclic peptide drug development.
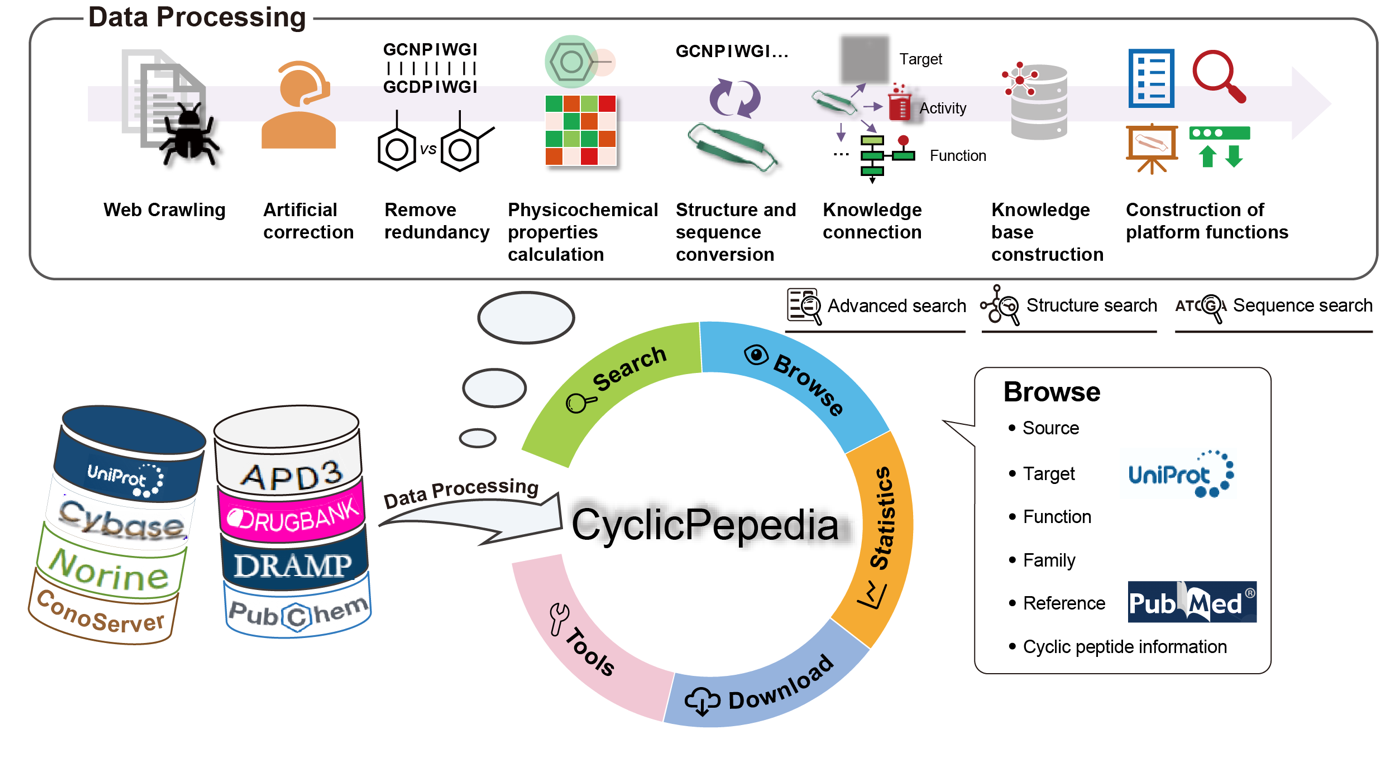
Browsing interface
CyclicPepedia provides multiple browsing interfaces dedicated to cyclic peptides, sources, functions, families, targets, references, and DrugBank. Click the Browse in the top navigation bar to enter different browsing interfaces.
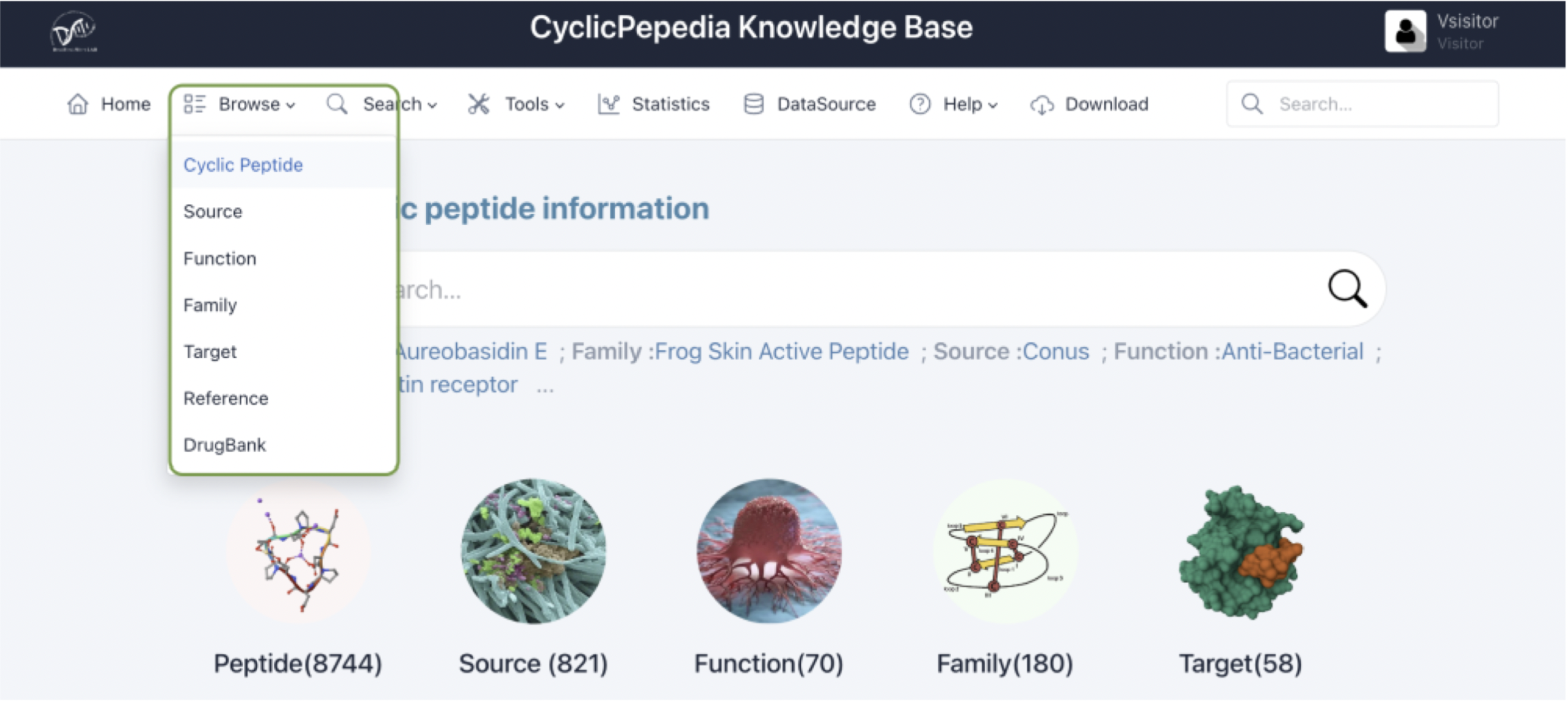
1) Cyclic peptide
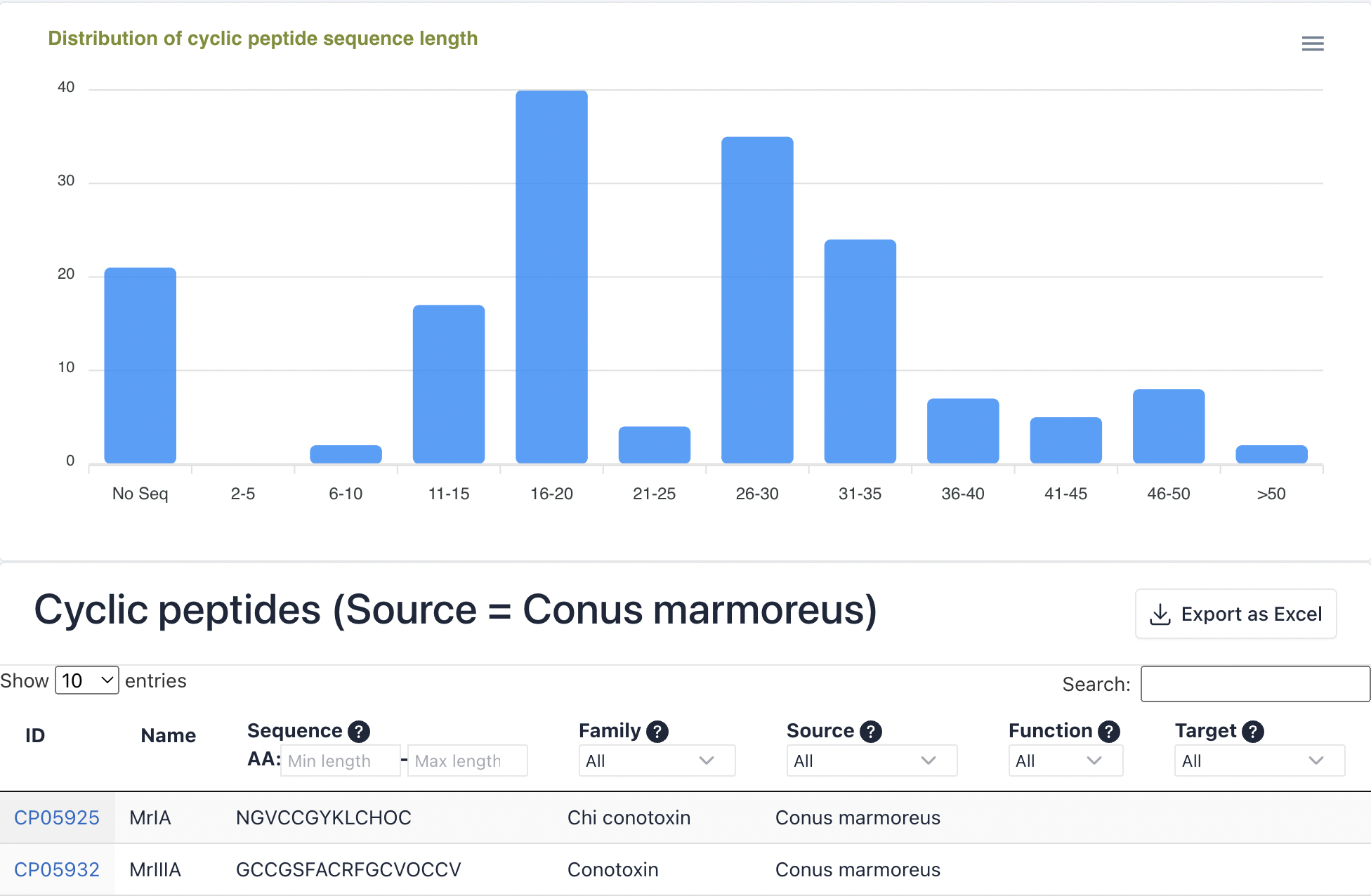
The cyclic peptide browsing interface consists of two parts: a bar plot displaying the distribution of cyclic peptide sequence length and a table of all cyclic peptides.

a. The bar plot can be downloaded through the button in the top-right corner.
b. Click the Export as Excel button to download the search results.
c. The number of cyclic peptide entries (i.e., 10, 25, 50, and 100) per page can be adjusted through the drop-down list.
d. Use the Search box to filter cyclic peptides by ID, name, sequence, families, sources, functions, and targets.
e. The Sequence, Family, Source, Function, and Target columns provide filtering functionality for the search results.
f. Click the CyclicPepedia ID to enter the corresponding cyclic peptide details page.
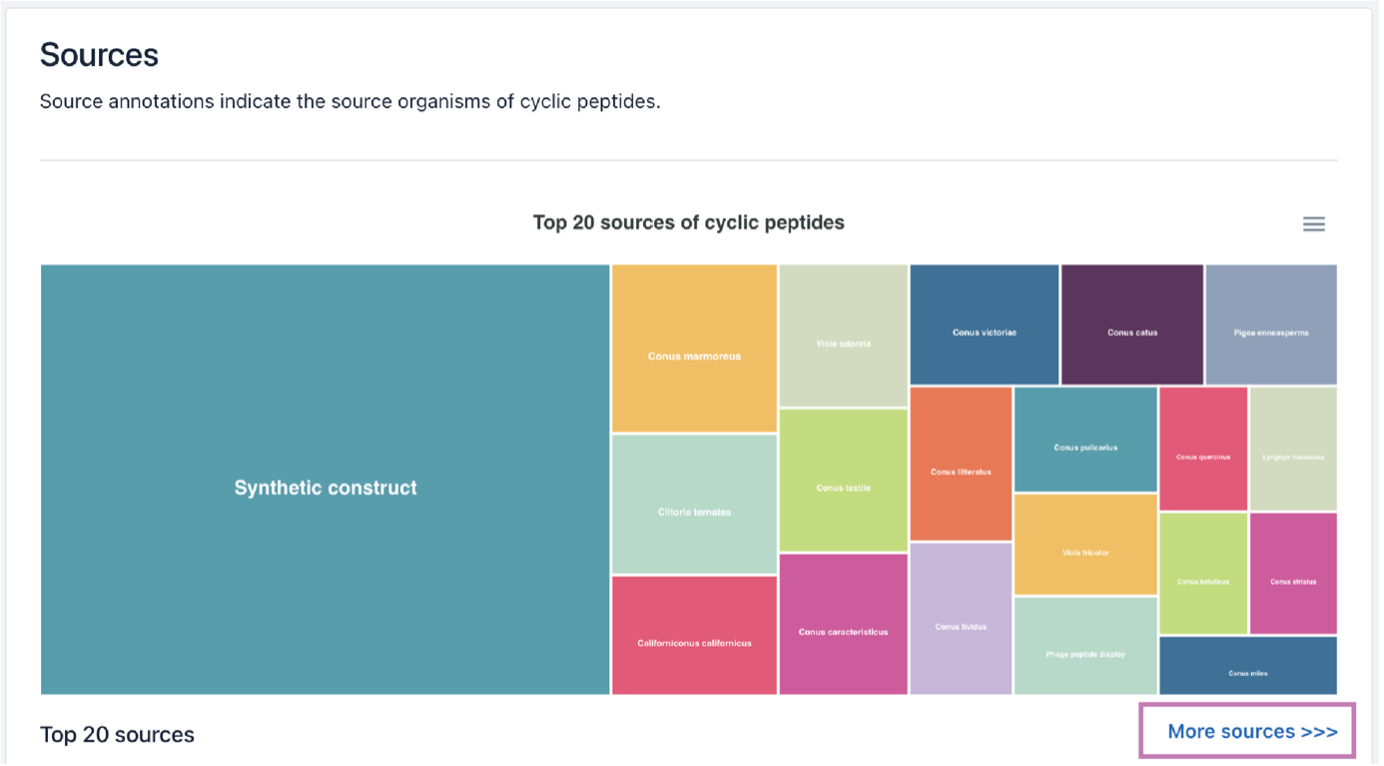
2) Source
The Source browsing interface displays the top 20 biological sources of cyclic peptides, and a full list of sources can be accessed by clicking the More sources button.
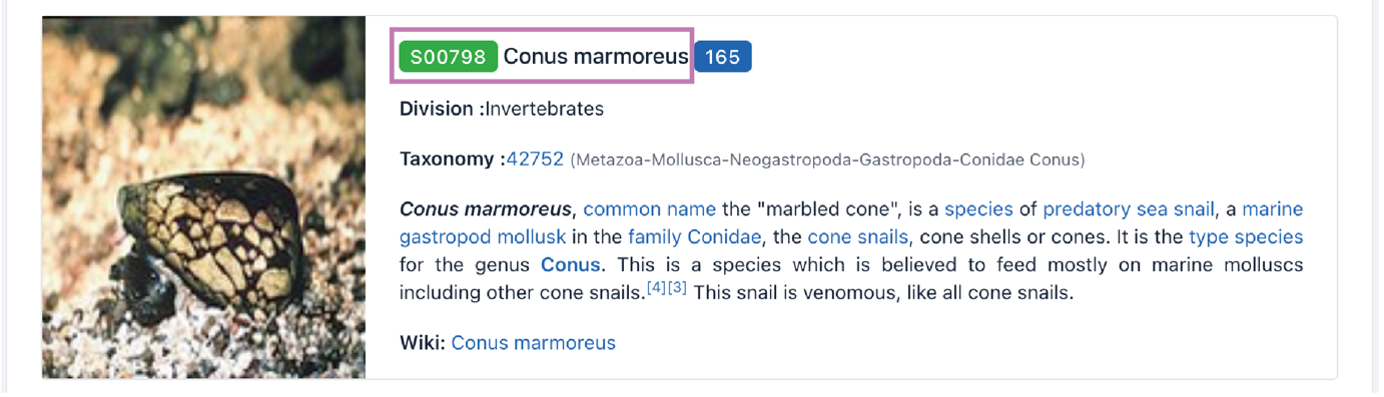
Click the source name to access the details page. The number of cyclic peptides included in this source classification is shown in the blue box. The Taxonomy ID, a general description, and a link to Wikipedia are listed.
The details page contains a statistical plot of cyclic peptide sequence length and a list of cyclic peptides derived from this source.
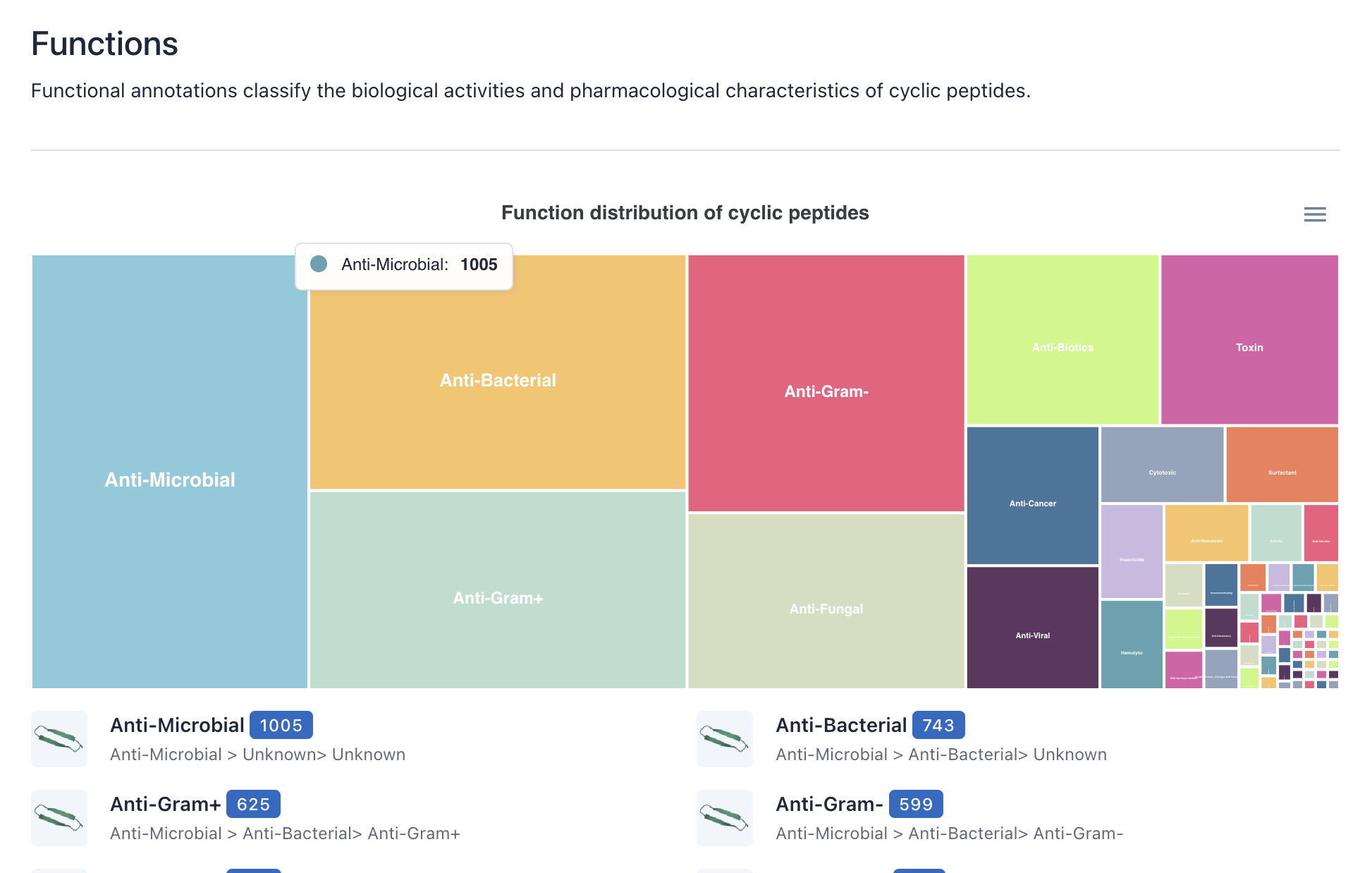
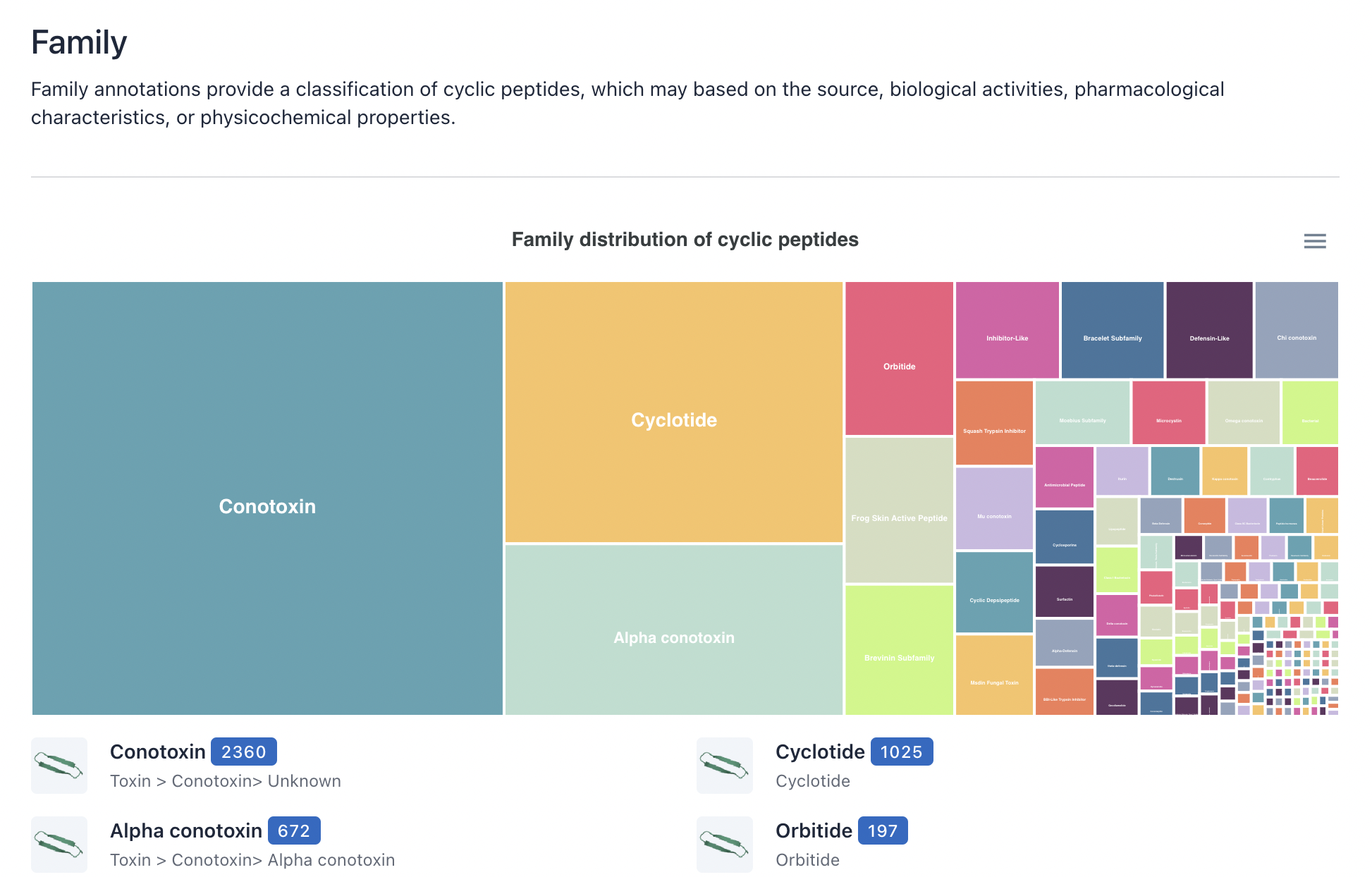
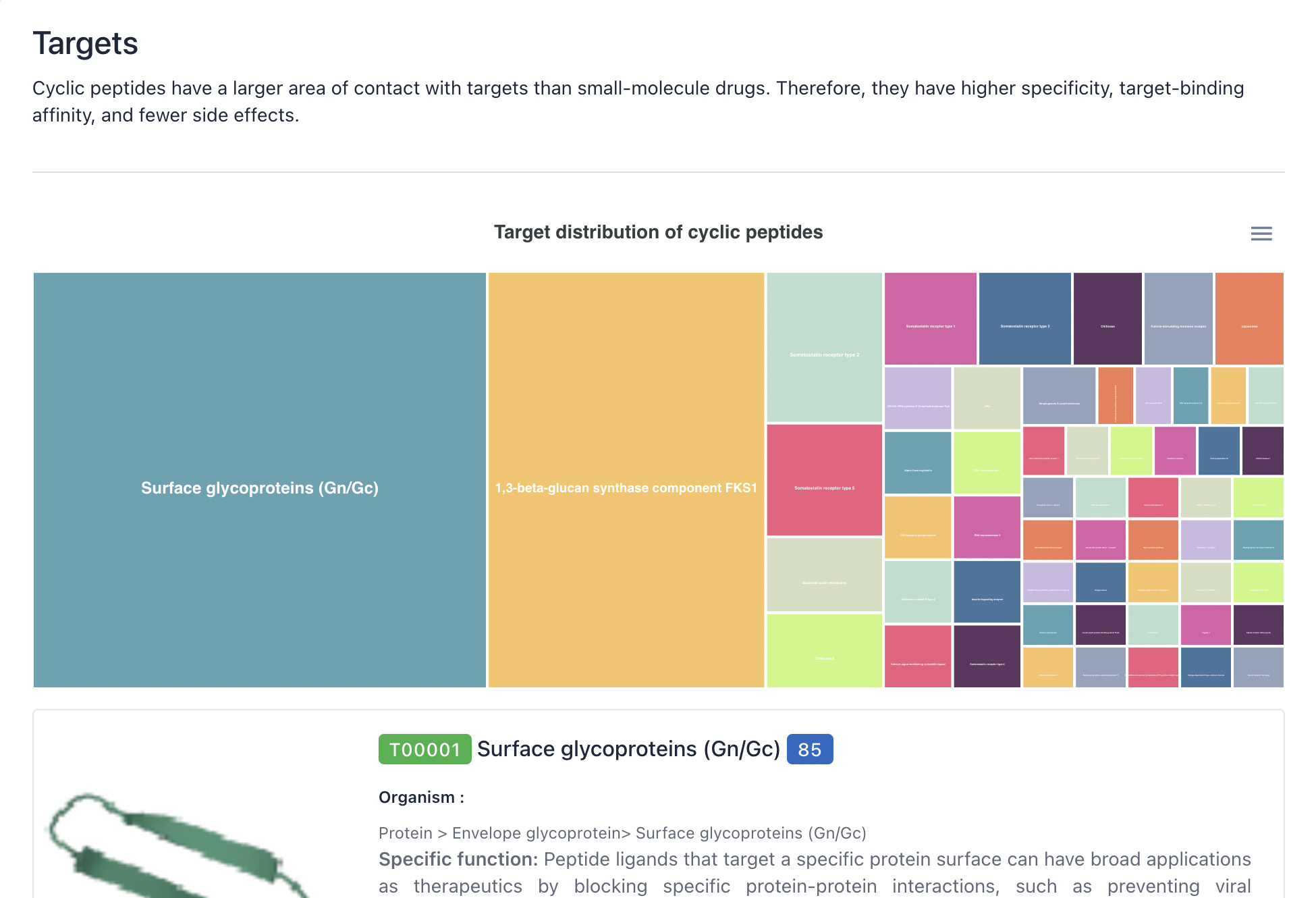
3) Function/ Family/ Target
The layout and functionality of the Function, Family, and Target browsing interfaces are similar to those of the Source browsing interface, please refer to the description above.
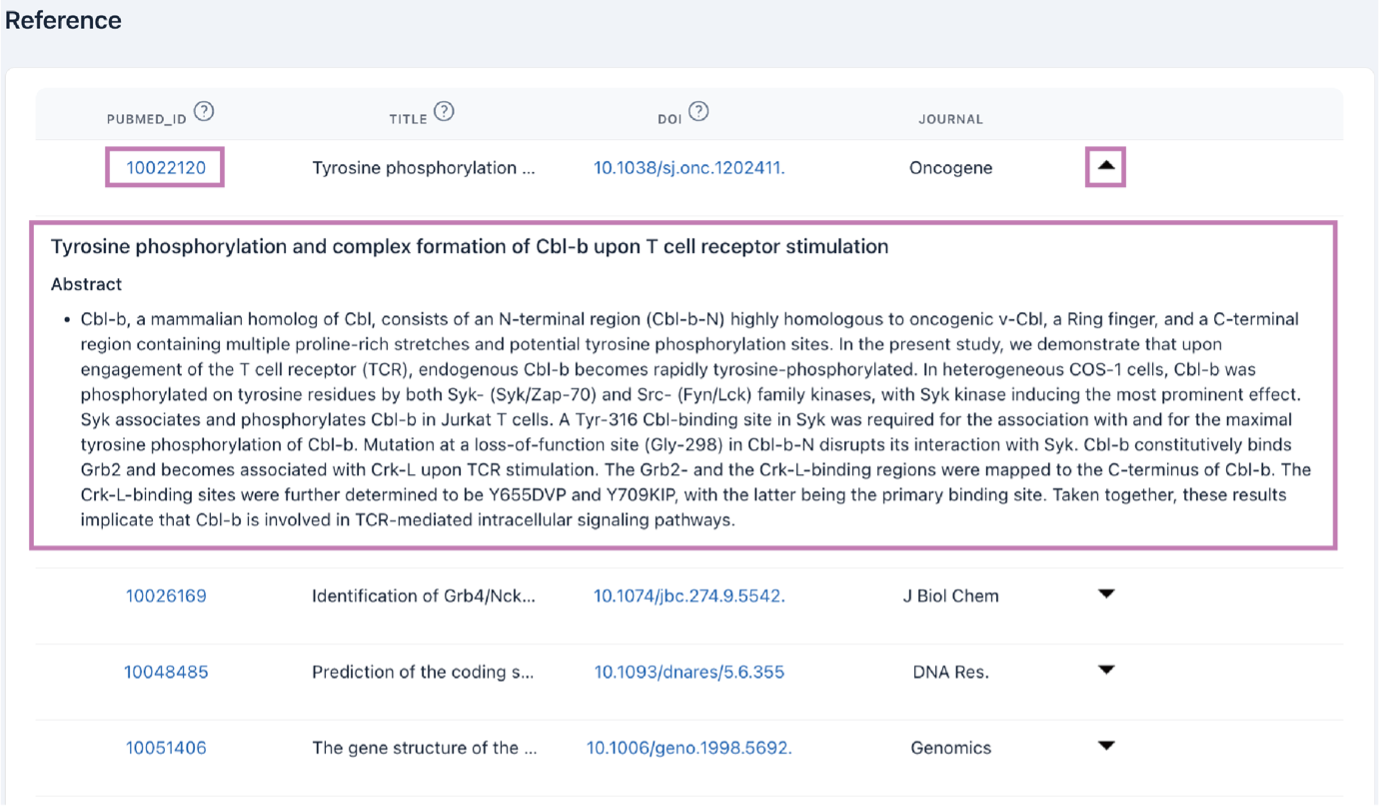
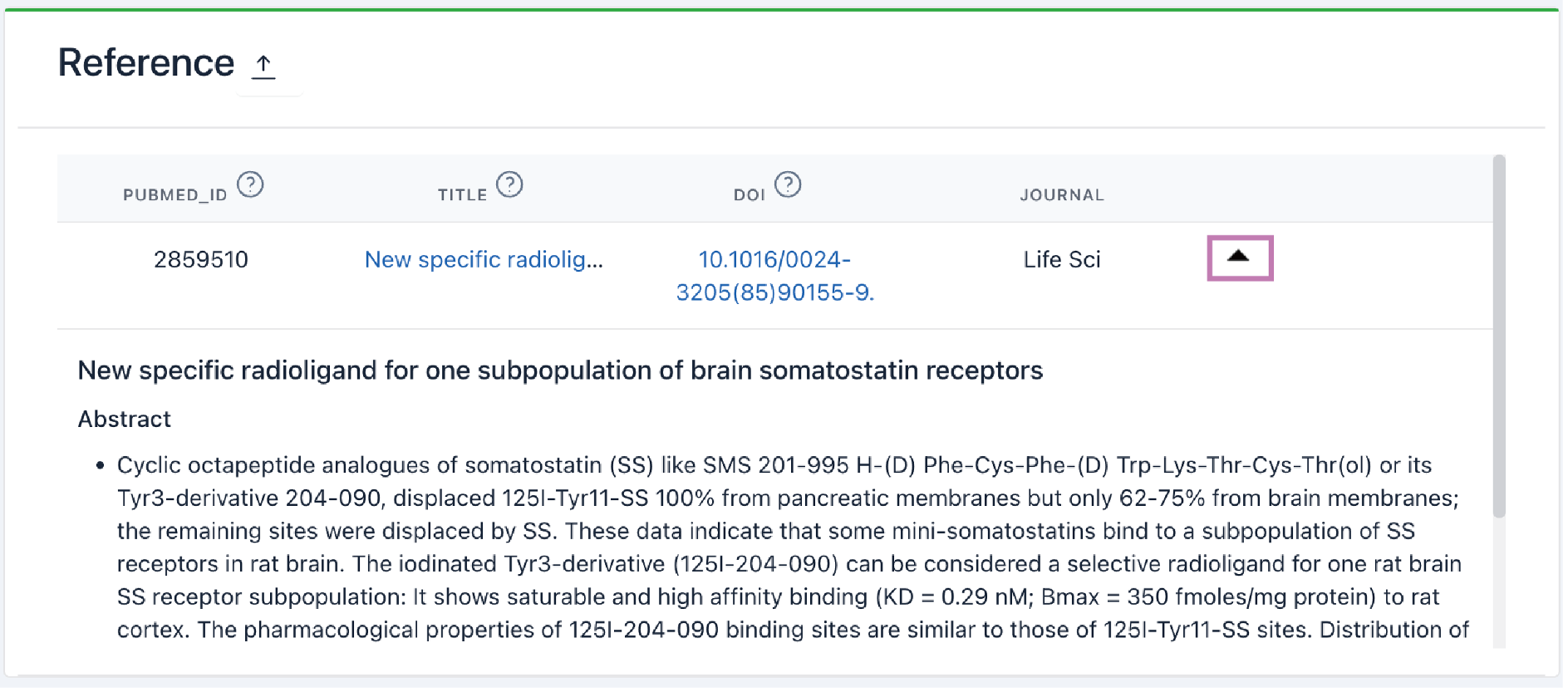
4) Reference
The reference browsing interface displays a full list of references with links to PubMed. Click the arrow to show abstracts.
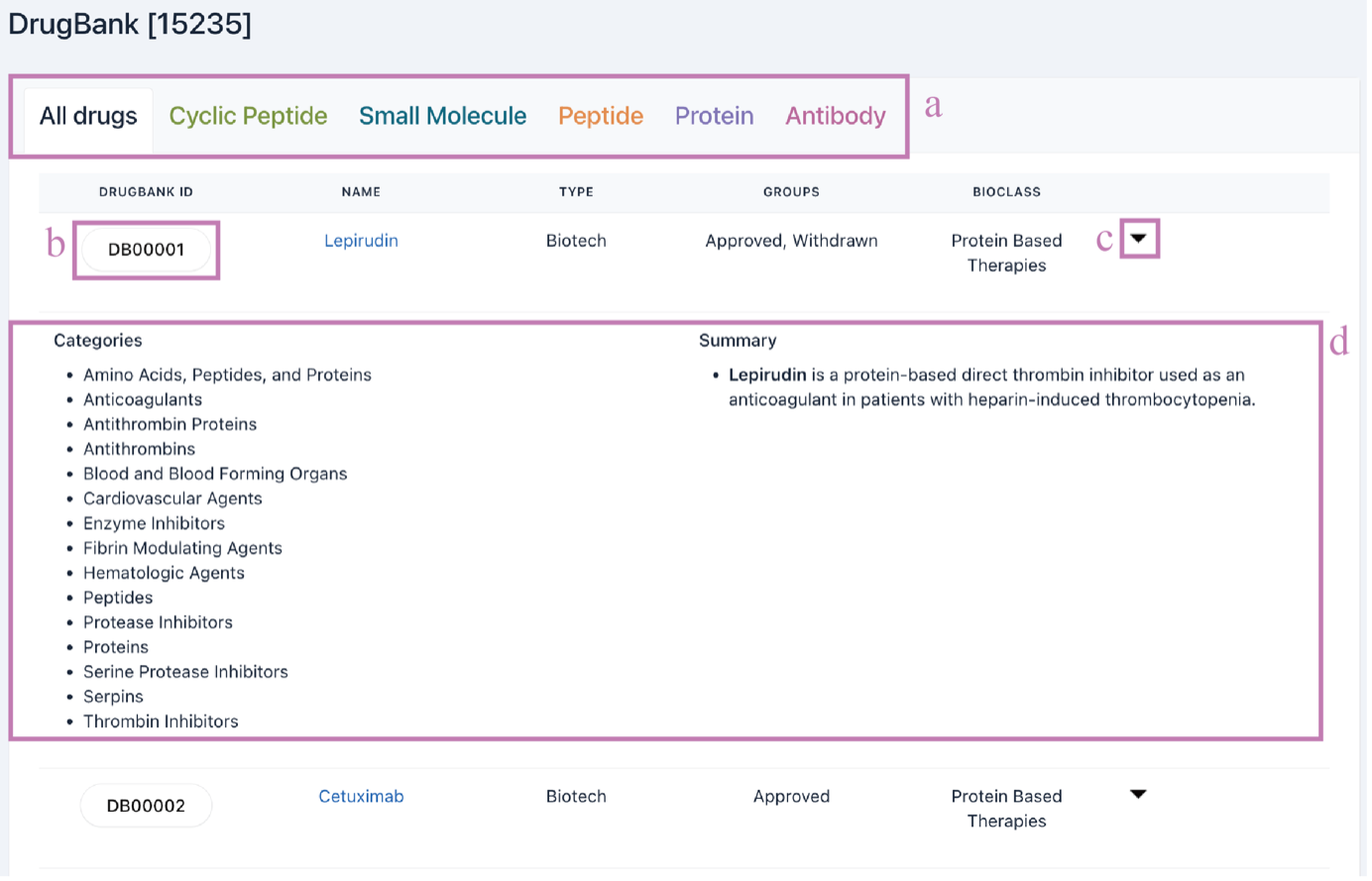
5) DrugBank
The DrugBank data is listed in the DrugBank browsing interface. Click the tab name (a) to show drugs according to their categories. Click the DrugBank ID (b) or the arrow (c) to show the drug summary (d).
Cyclic peptide details page
The cyclic peptide details page contains basic information, structure, sequence, biologic determination, chemical and physical properties, binding target, manufacturers, forecasting tools, information sources, and references. These data can be quickly accessed through the navigation bar (a) on the left.

Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060
1) Basic information: This section displays CyclicPepedia ID, IUPAC name, synonyms, source, family, function, description information, and a knowledge network.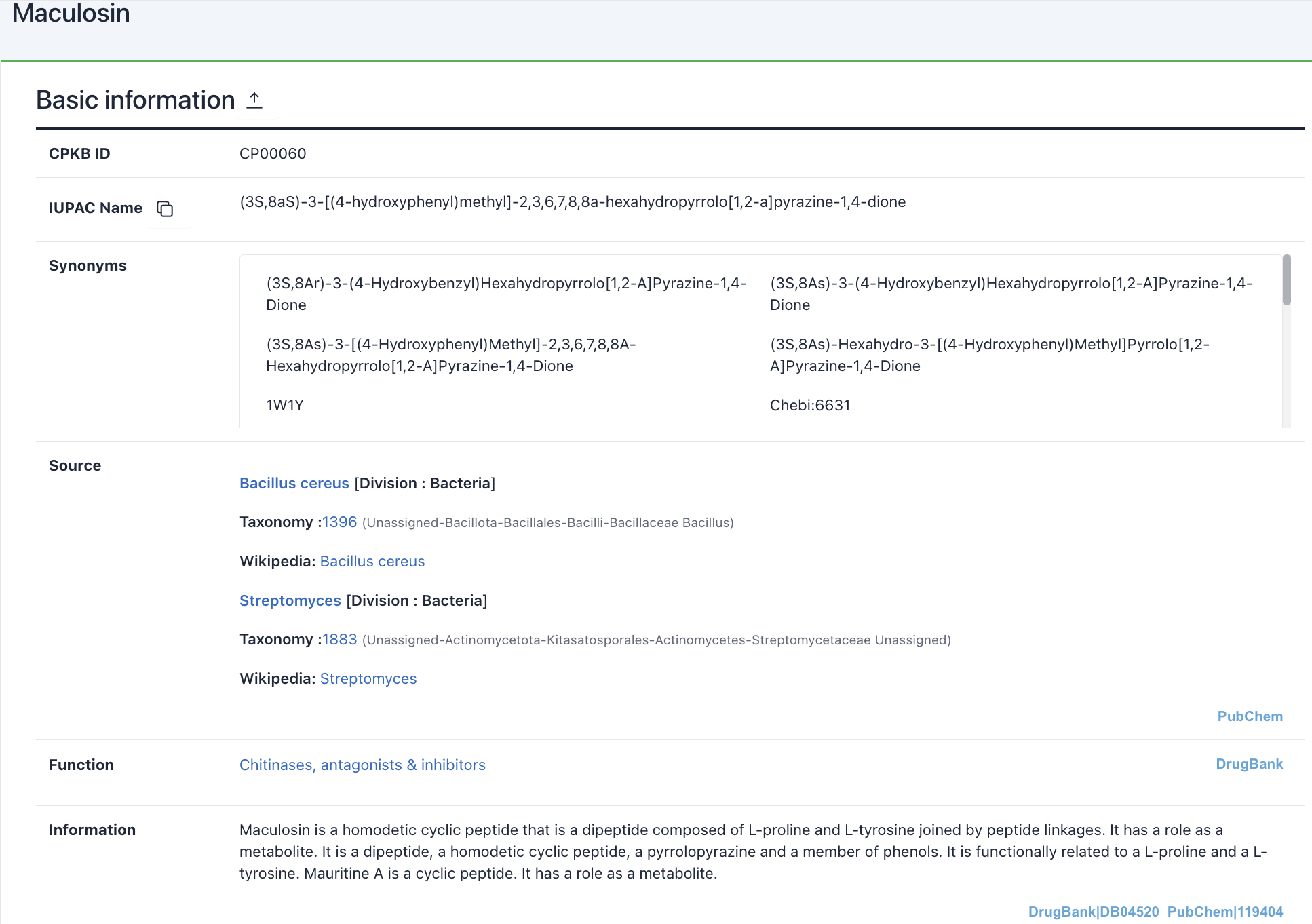
Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060
Click the network node to enter the cyclic peptide details page or the corresponding external database.
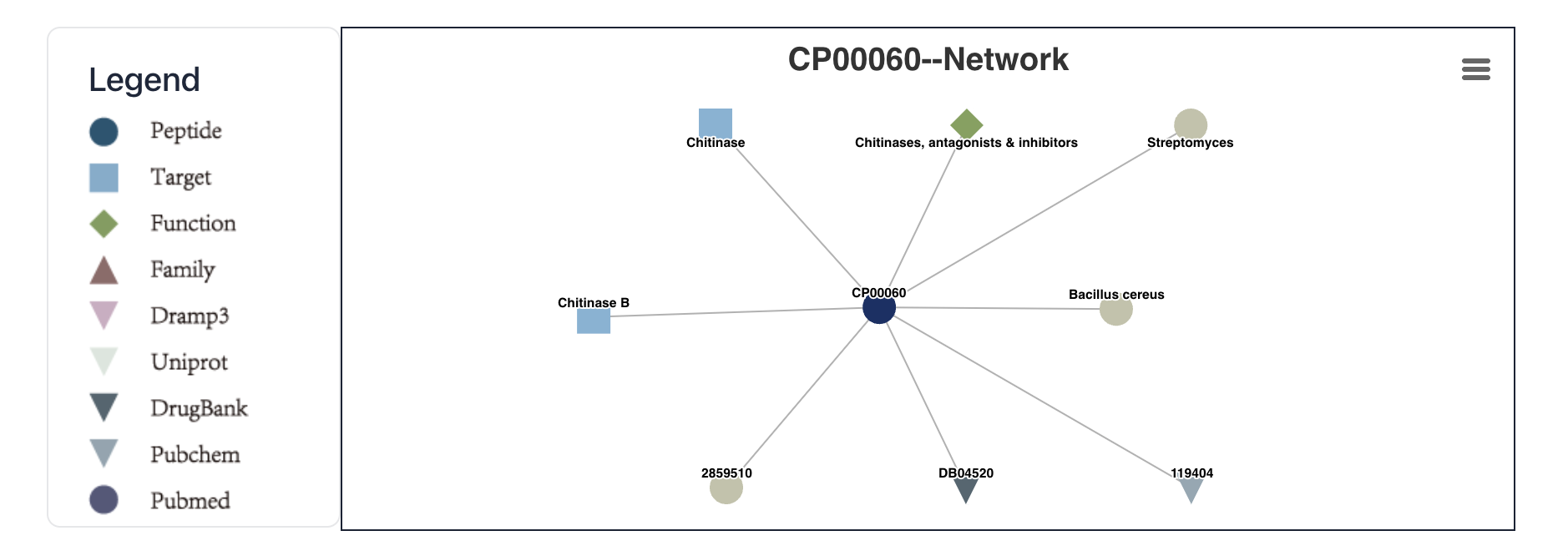
Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060
2) Structure: This section provides data on cyclic peptide molecular formula, molecular weight, SMILES, InchI, InChI Key, and two-dimensional and three-dimensional structures.
Click the similarity structure (a) button to search for cyclic peptides with similarity scores > 0.9. Structure files can be downloaded by clicking the download (b) button.

Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060#Structure
The sources of structural data and the types of 3D structures (e.g., complex) are listed on the right.
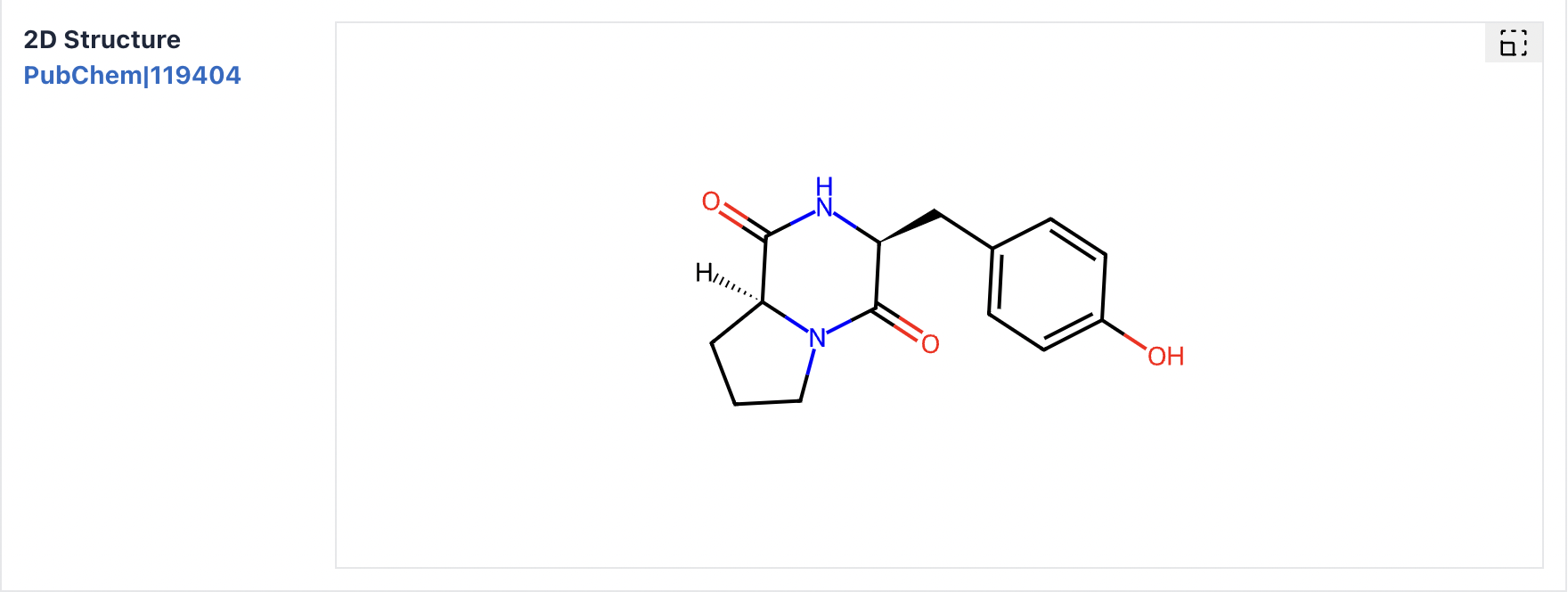
Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060#Structure
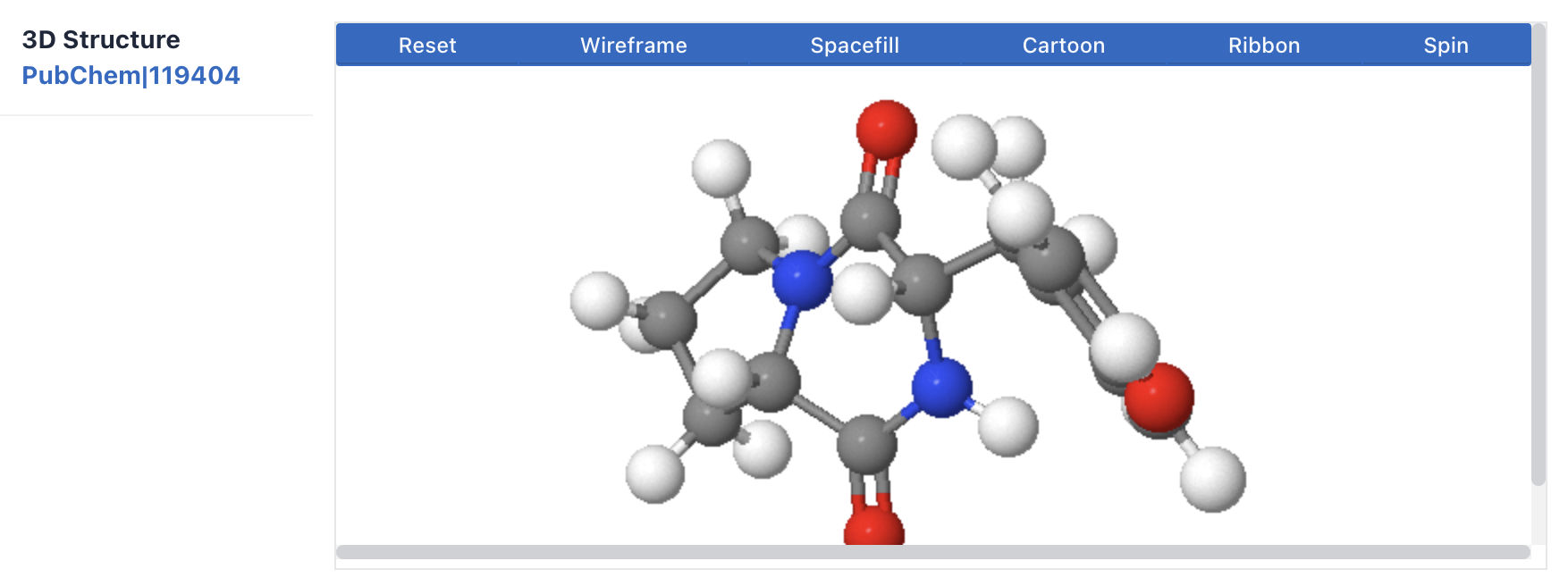
Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060#Structure
Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060#Structure
3) Sequence: This section presents different sequence formats, for example, one-letter code, IUPAC condensed, amino acid chain, graph representation, and SVG image, as well as a plot of amino acid composition and a report of the Structure-to-Sequence (Struc2Seq) transformation.
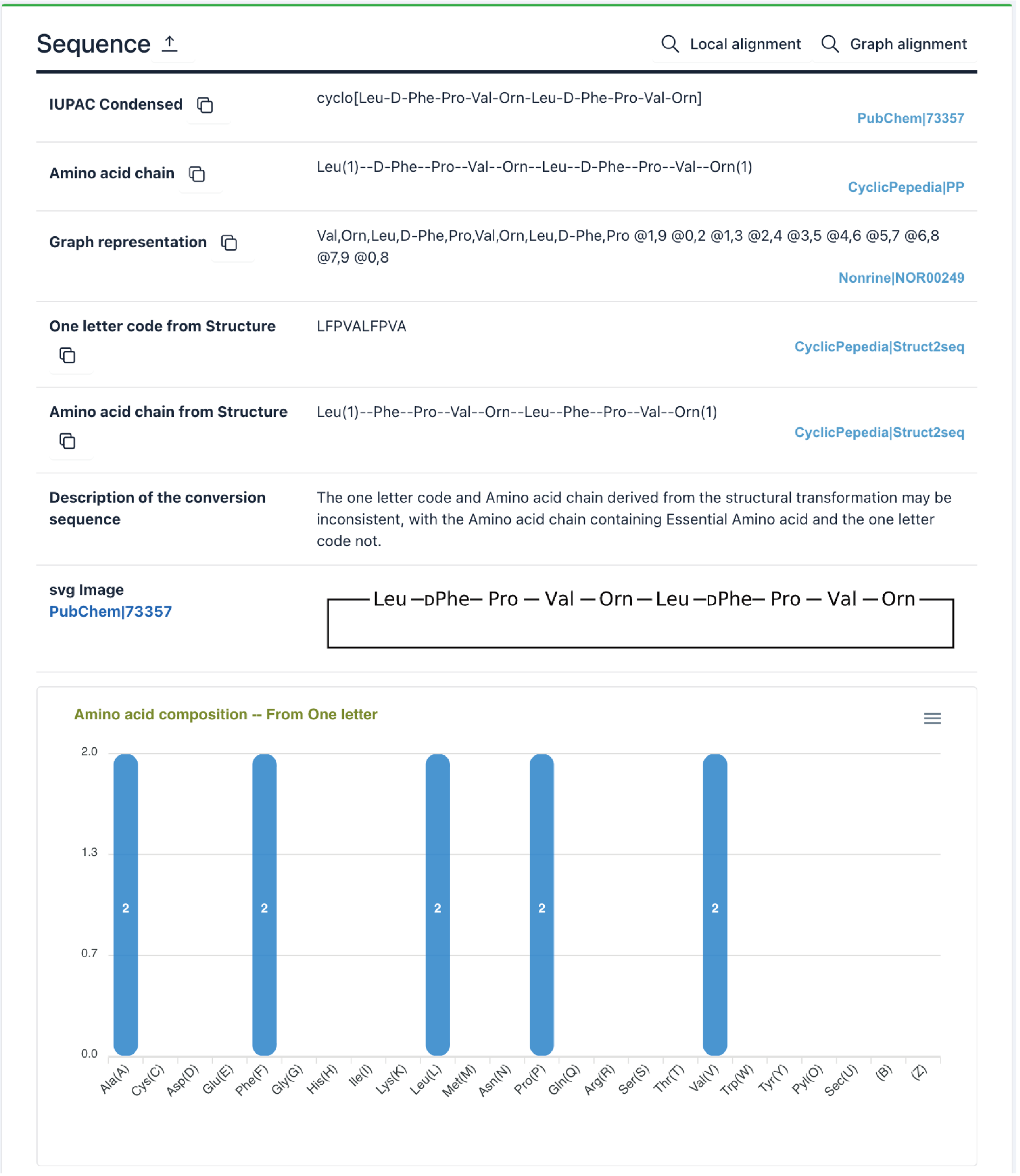
Gramicidin S: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00038#Sequence
Refer to the Tools|Structure to Sequence for a description of the Struc2Seq report.

Gramicidin S: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00038#Sequence
Click the Local alignment or Graph alignment button to query similar sequences. Refer to the Search|Sequence Search for details.

4) Biologic Determination: This section lists bioassay results related to the cyclic peptide.
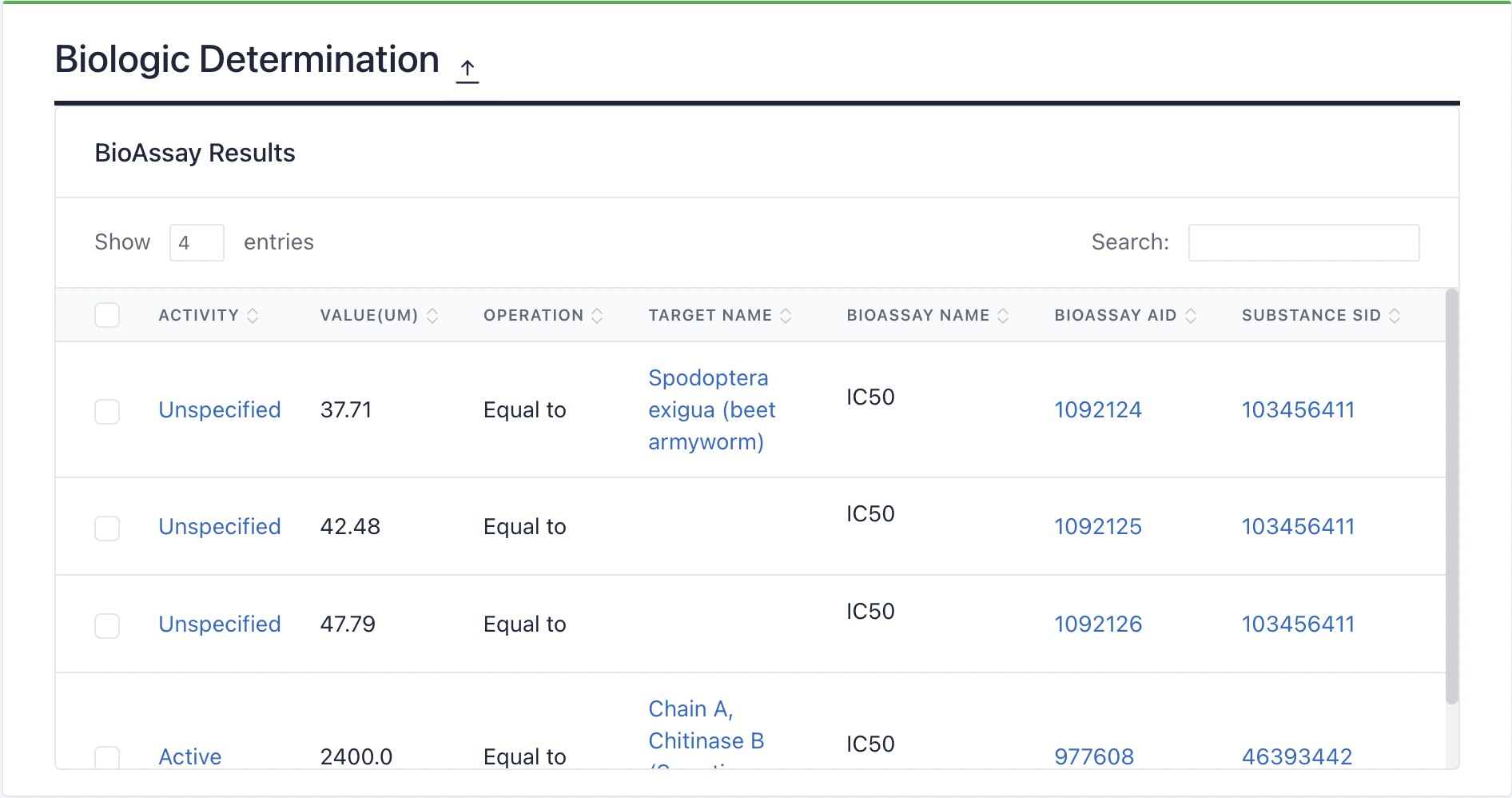
Maculosin: https://www.biosino.org/iMAC/cyclicpepedia/detail?id=CP00060#Biologic%20Determination
5) Chemical and Physical Properties: It contains two parts—the structural properties and the sequence properties computed by CyclicPepedia.
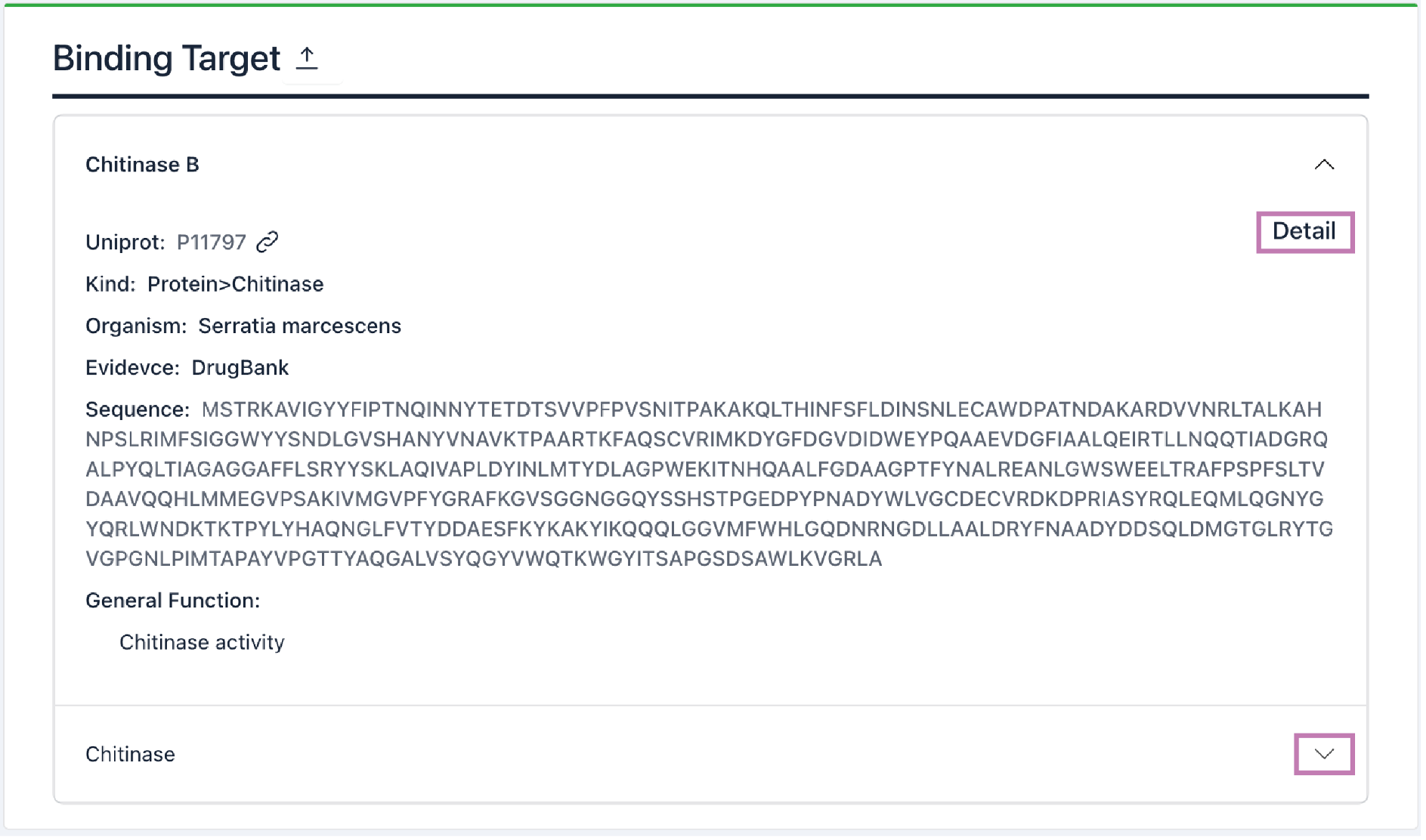
6) Binding Target: Data on associated targets are presented in this section. Click the arrow button to expand/collapse the information tab. Click the Detail button to access the target's details page.
7) Manufacturers: CyclicPepedia provides links to popular manufacturers such as Merck, Baxter Healthcare Corp, and Upsher-Smith laboratories.
8) Forecasting tools: It presents connections to CyclicPepedia tools such as Structure-to-Sequence conversion, Sequence-to-Structure conversion, Structure Properties, and Sequence Properties computation, as well as several external predictive tools.
9) Information Source: This section lists links to external information sources.
10) Reference: This section lists associated literature with links to PubMed. Click the arrow to show abstracts.
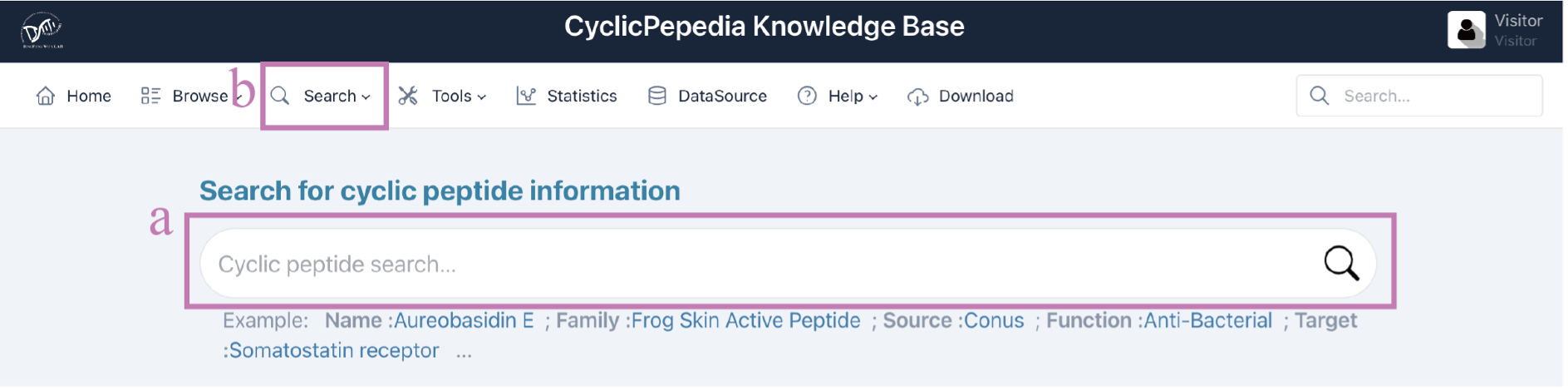
Search tools
CyclicPepedia offers four search methods: quick full-text search (a) on the homepage, advanced search, structure search, and sequence search. Click the Search (b) to select search methods.
1) Advanced search
Click Search|Advanced Search to enter the Advanced Search page.
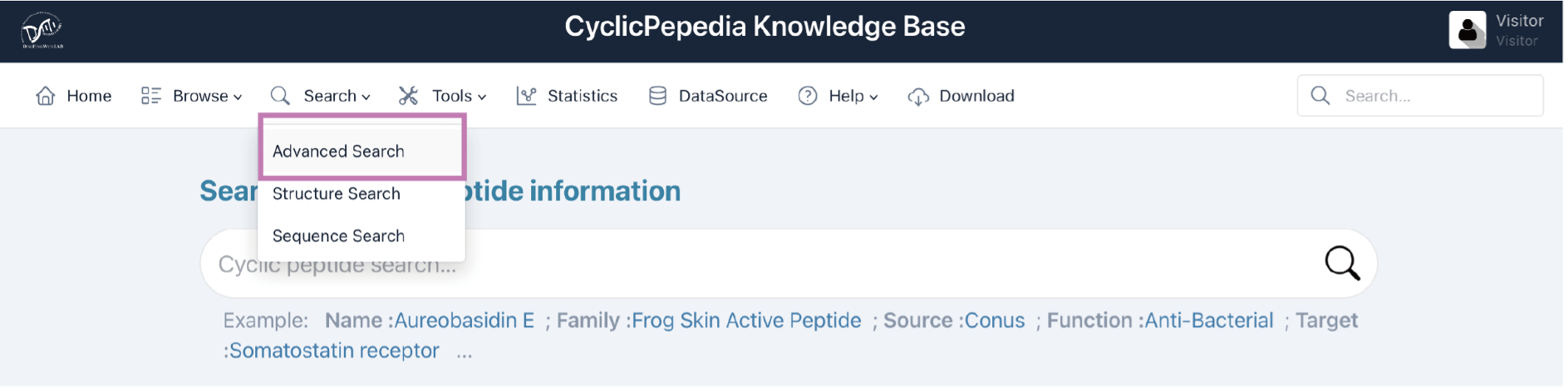
Advanced search provides users with multiple criteria, for example, sequence and structure information, physiochemical properties, sequence properties, and biological annotation information, to create custom search queries. Click the arrow button to expand the filtering criteria.
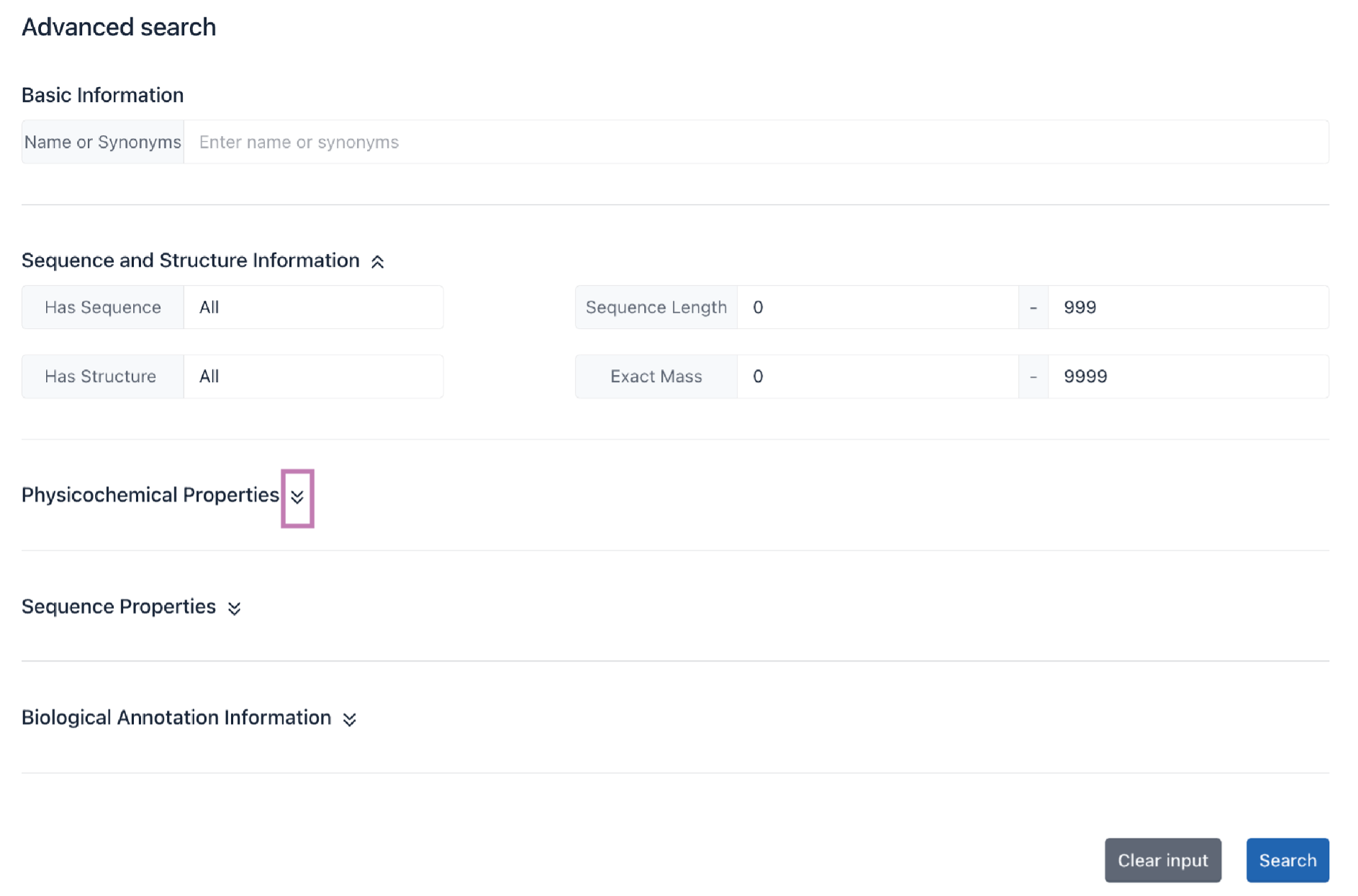
Enter the cyclic peptide name and/or select filtering criteria, and click the Search button to get search results. Click the Clear input button to clear all inputs.
Example: We want to search for anti-bacterial cyclic peptides that have sequence and structure information and have amino acid sequence length > 5. The filtering criteria are: "Has Sequence = Yes” & “Sequence Length > 5" & "Has Structure = Yes” & "Function = Anti-Bacterial (743)"
Click the Search button.
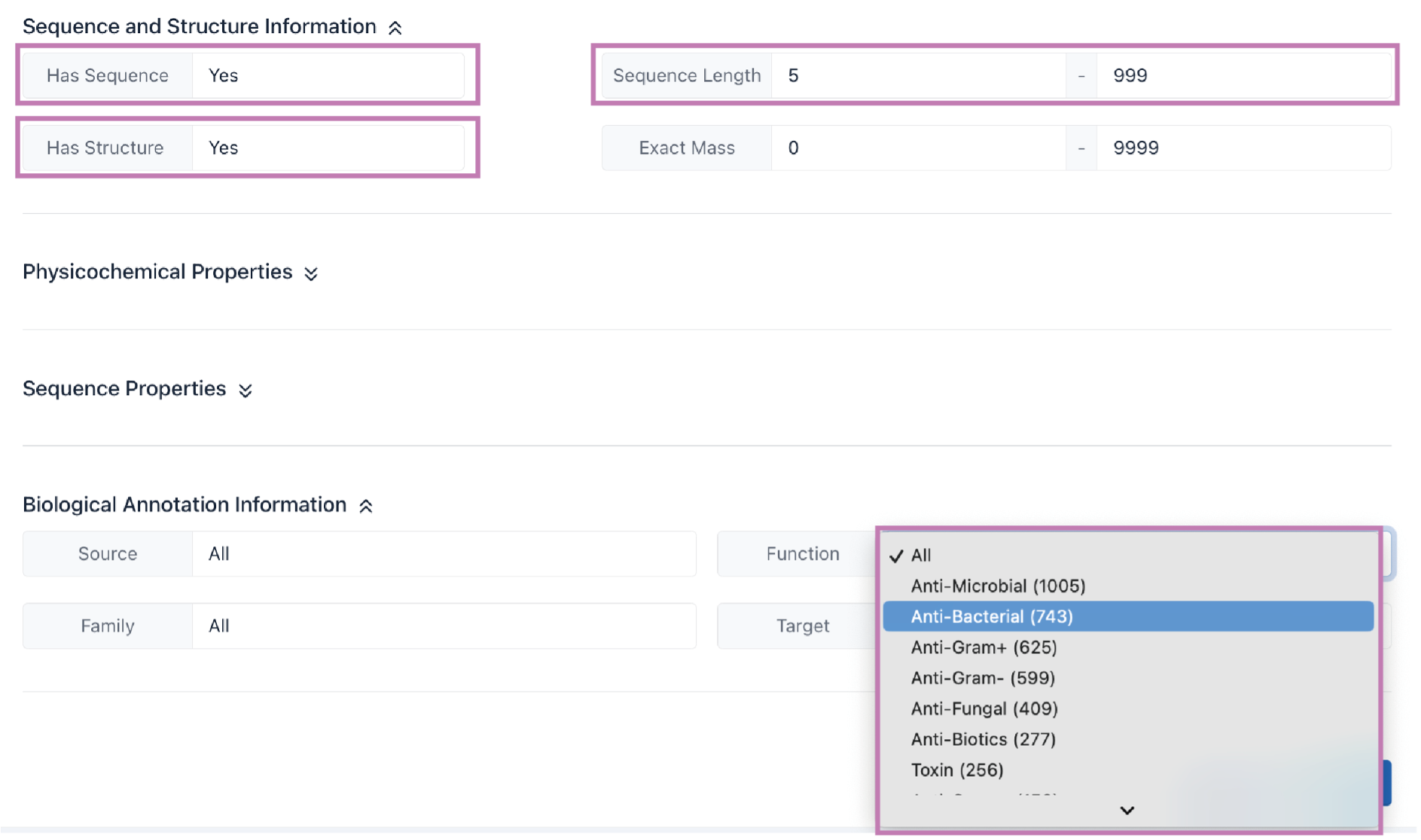
The Search result table is presented at the bottom of the page.
a. Click the Export as Excel button to download the search results.
b. The number of cyclic peptide entries (i.e., 10, 25, 50, and 100) per page can be adjusted through the drop-down list.
c. Use the Search box to filter search results by ID, name, sequence, families, sources, functions, and targets.
d. The Sequence, Family, Source, Function, and Target columns provide filtering functionality for the search results.
e. Click the CyclicPepedia ID to enter the corresponding cyclic peptide details page.
2) Structure search
Click Search|Structure Search to enter the Structure Search page.
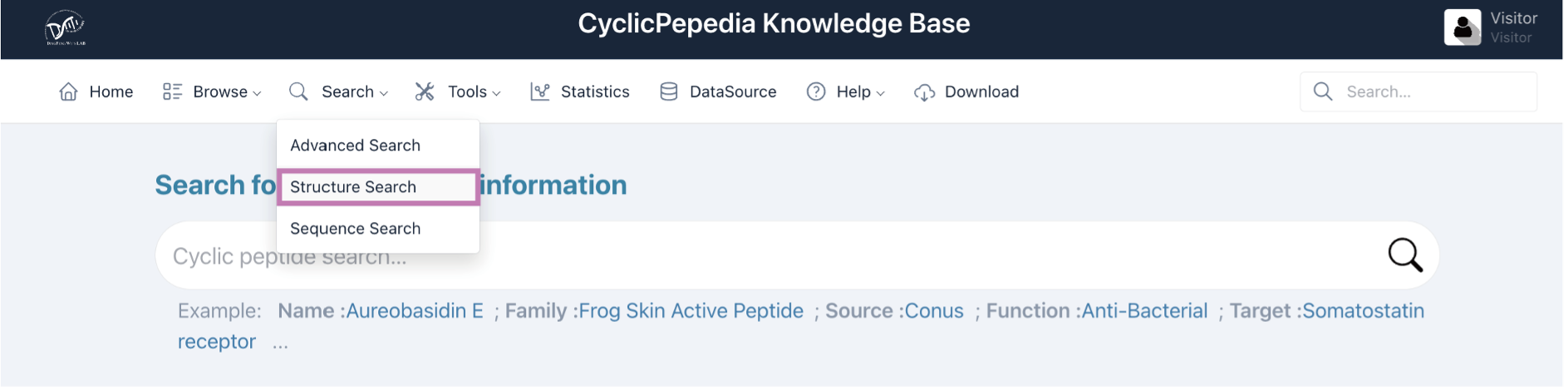
Tip! Structure search may take a long time, depending on input molecules and parameter selection.
Step 1. Upload structure files in PDB/SDF formats or paste your SMILES into the left panel.
Example: CC1C(C(=O)NC(C(=O)N2CCCC2C(=O)N(CC(=O)N(C(C(=O)O1)C(C)C)C)C)C(C)C)NC(=O)C3=C4C(=C(C=C3)C)OC5=C(C(=O)C(=C(C5=N4)C(=O)NC6C(OC(=O)C(N(C(=O)CN(C(=O)C7CCCN7C(=O)C(NC6=O)C(C)C)C)C)C(C)C)C)N)C
Step 2. Select search type, for example, exact match, substructure search, and similarity search.
Step 3. For similarity search, users can choose the molecular fingerprint types and the similarity metrics. The fingerprint types include RDKit Fingerprint, MACCS Keys (Molecular ACCess System), and Morgan Fingerprint; The similarity metrics include Tanimoto similarity and Dice similarity.
Step 4. Adjust the slider to select the Similarity threshold range.
Step 5. Click the Search button to get search results.
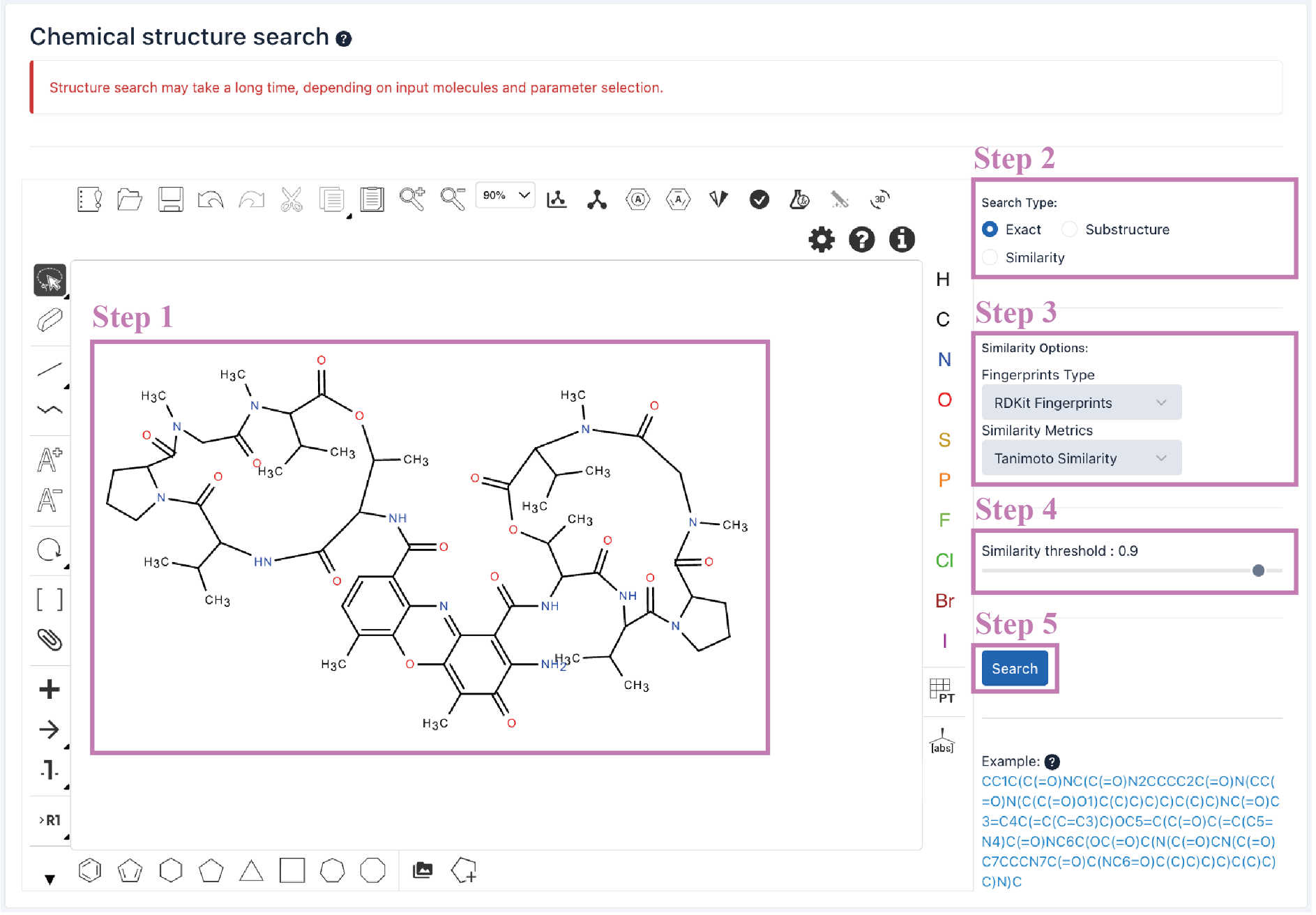
The Search result table is presented at the bottom of the page.
a. Click the Export as Excel button to download the search results.
b. The number of cyclic peptide entries (i.e., 10, 25, 50, and 100) per page can be adjusted through the drop-down list.
c. Use the Search box to filter search results by ID, name, molecular formula, and similarity score.
d. Click the CyclicPepedia ID to enter the corresponding cyclic peptide details page.

3) Sequence search
Click Search|Sequence Search to enter the Sequence Search page. The sequence search is divided into local alignment and graph alignment.
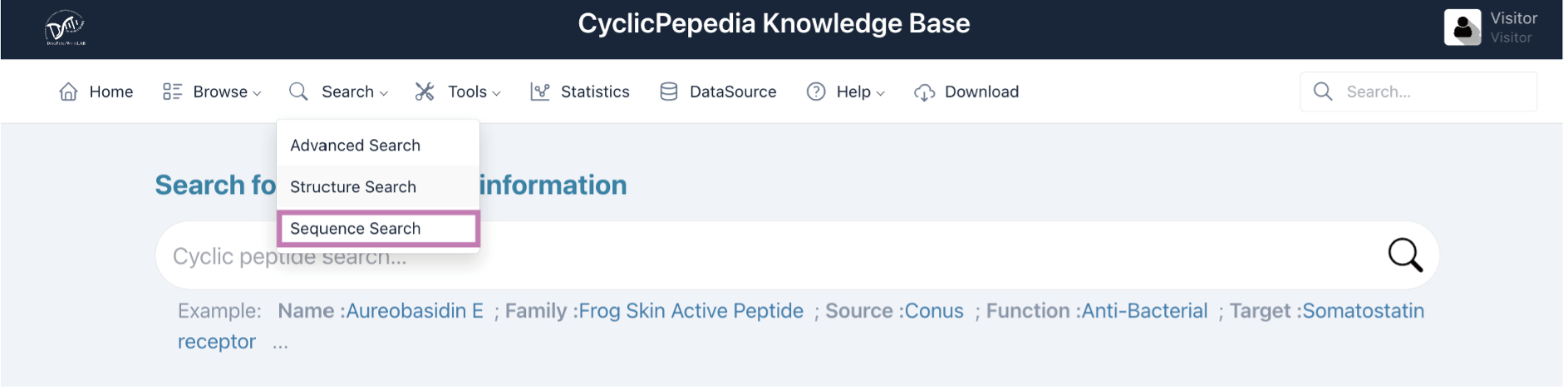
i. Local alignment
The Smith-Waterman algorithm is utilized for the Local alignment of peptide sequences (Biopython: pairwise2.align.localxx, https://biopython.org/)..) This method allows for parameter adjustment to set penalties for matches, mismatches, and gaps, ensuring tailored alignment based on specific sequence characteristics.
Tip! This method is only applicable to peptides represented by one-letter amino acid codes.
Step 1. Enter or paste your peptide sequence into the panel.
Example:FLPAVIRVAANVLPTAFCAISKKC
Step 2. Adjust the algorithm parameters, for example, match score, mismatch score, open gap score, and extend gap score, to set penalties for matches, mismatches, and gaps; The e-value threshold is used to filter the alignment results.
Step 3. Click the Search button to perform the local alignment.
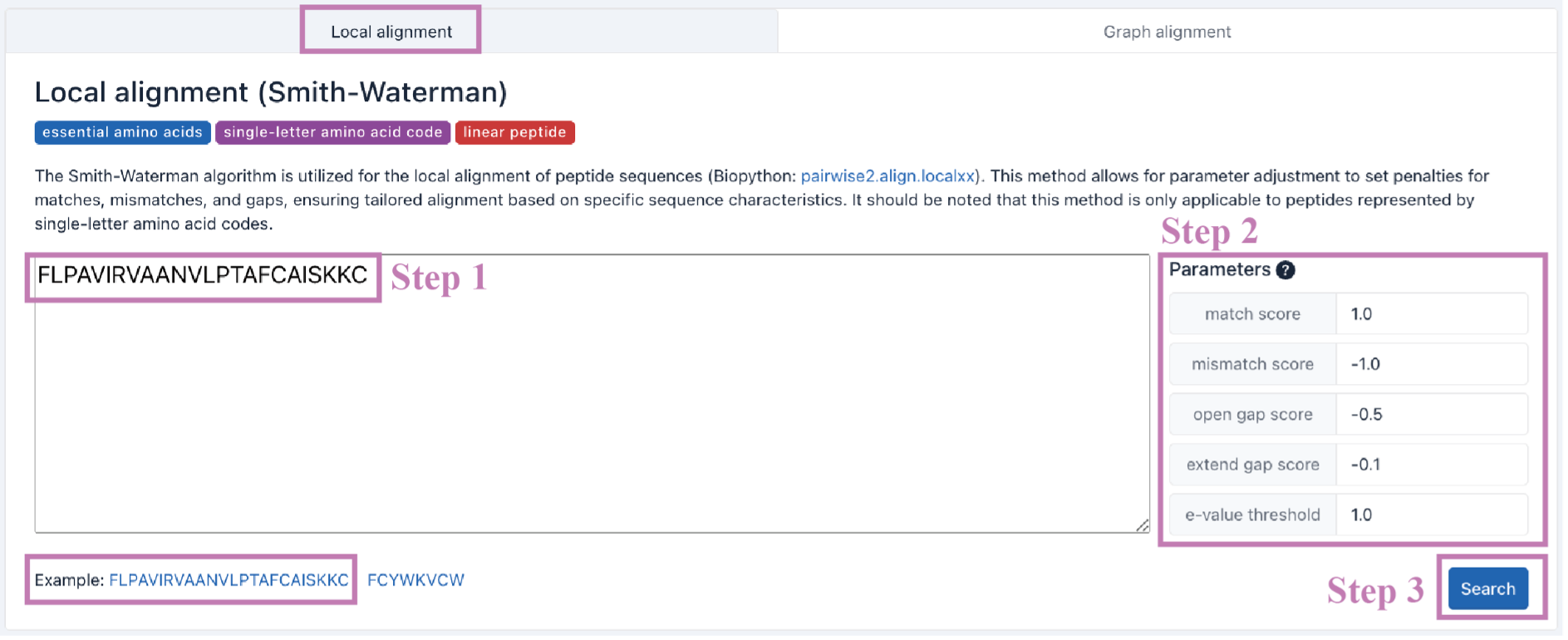
The Search result table is presented as a five-column table.
ID: CyclicPepedia ID.
Name:cyclic peptide name.
Sequence: peptide sequence.
Score: the alignment score.
E-value: the expected value. A measure of the significance of the match.
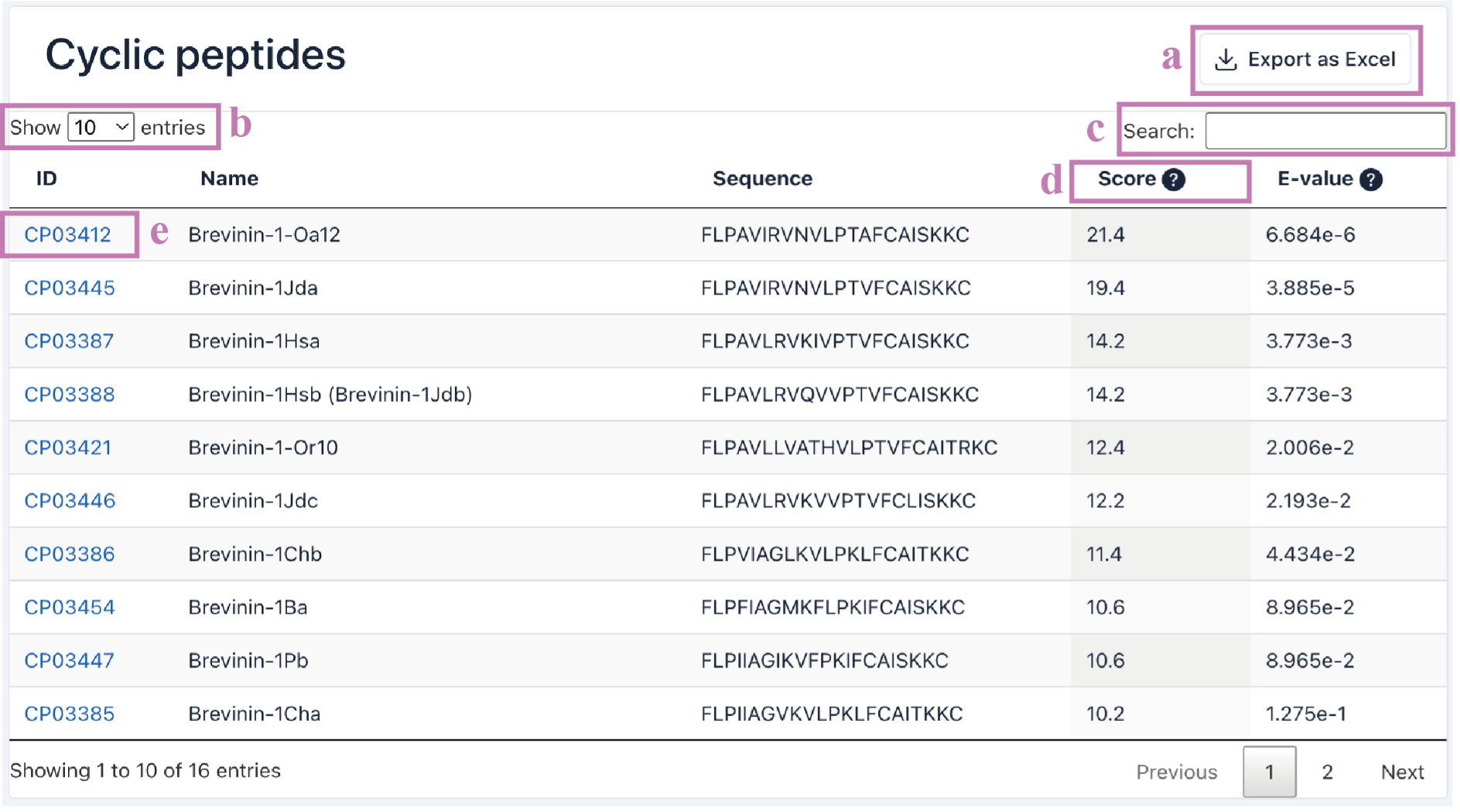
a. Click the Export as Excel button to download the search results.
b. The number of cyclic peptide entries (i.e., 10, 25, 50, and 100) per page can be adjusted through the drop-down list.
c. Use the Search box to filter search results by ID, name, sequence, alignment score, and E-value.
d. Click the column name to sort the results.
e. Click the CyclicPepedia ID to enter the corresponding cyclic peptide details page.
ii. Graph alignment
To leverage the cyclization information in cyclic peptide sequences, we developed a graph alignment algorithm based on NetworkX. The graph alignment can convert cyclic peptides into graphical structures and measure the similarity by Graph Isomorphism. The input sequence format can be IUPAC condensed, amino acid chain, and graph presentation (refer to Sequence format transformation for sequence examples).
Tip! Users can adjust parameters to reduce search space (e.g., set min AA <= sequence length <= max AA and set a higher value for AA comp threshold).
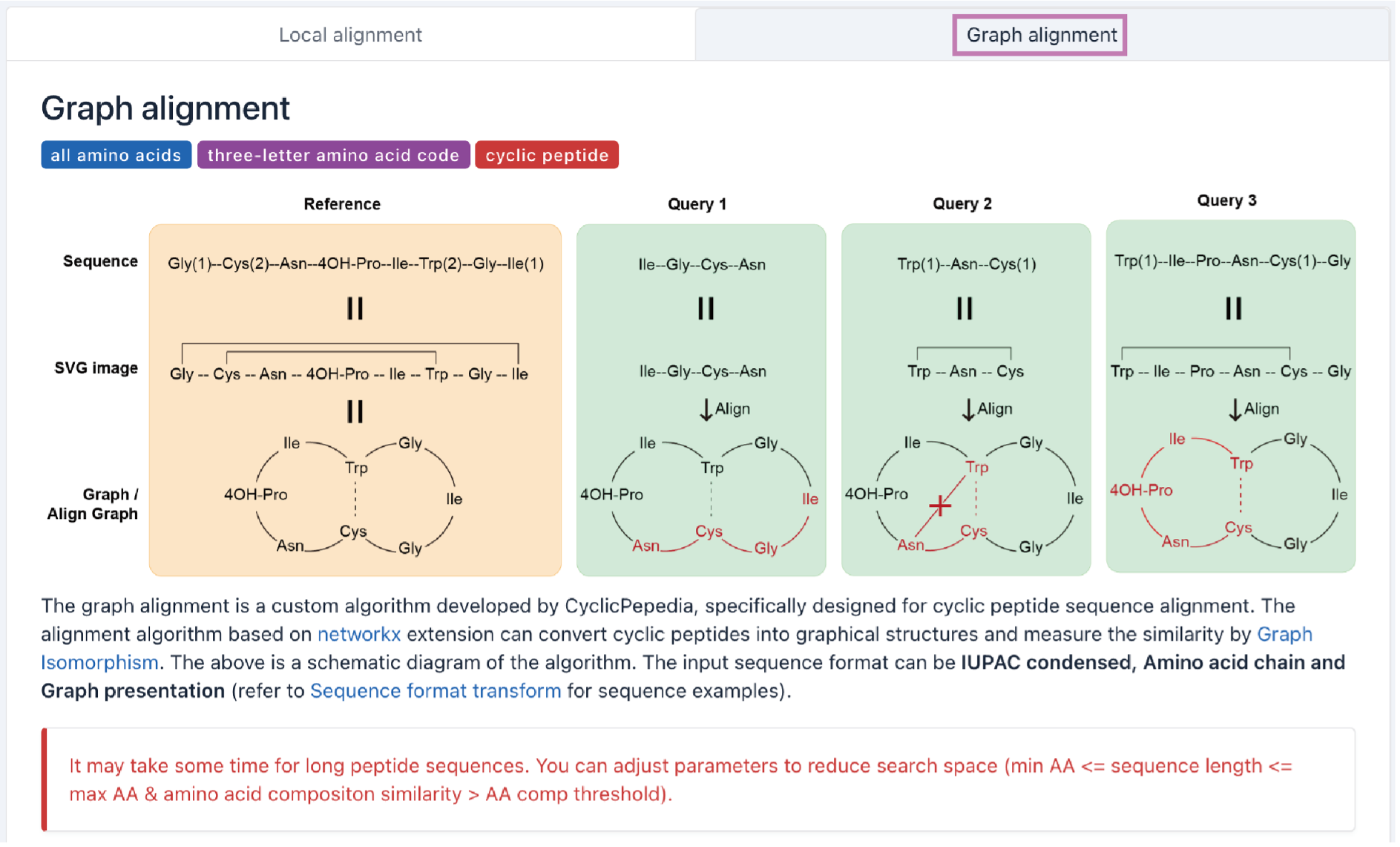
Step 1. Enter or paste your sequence into the panel.
Example: Cys(1)--Tyr--Trp--Lys--Val--Cys(1)
Step 2. Adjust the algorithm parameters.
min AA: minimum amino acid sequence length.
max AA: maximum amino acid sequence length.
AA comp threshold: threshold for amino acid composition similarity.
Similarity threshold: threshold for filtering alignment results.
Step 3. Click the Search button to perform the graph alignment.

The Search result table is presented as a five-column table.
ID: CyclicPepedia ID.
Name:cyclic peptide name.
Sequence: peptide sequence.
Matched: the number of matched graph nodes and edges.
Similarity: graph similarity, is the degree of similarity between nodes or edges in a network.

a. Click the Export as Excel button to download the search results.
b. The number of cyclic peptide entries (i.e., 10, 25, 50, and 100) per page can be adjusted through the drop-down list.
c. Use the Search box to filter search results by ID, name, sequence, matched, and similarity.
d. Click the column name to sort the results.
e. Click the CyclicPepedia ID to enter the corresponding cyclic peptide details page.
Cyclic peptide tools
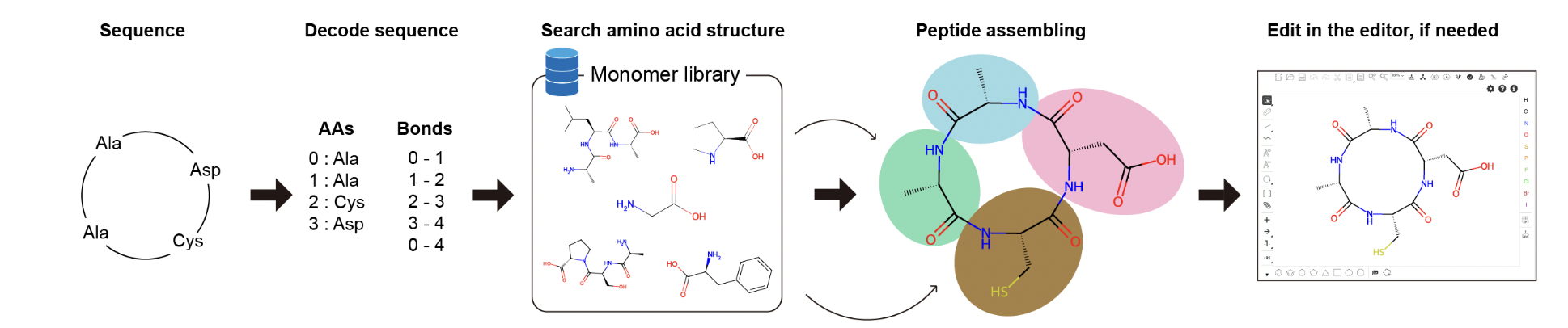
1) Structure to sequence
Click the Tools|Structure to Sequence to enter the Structure-to-Sequence conversion page.
Structure-to-Sequence (Struc2Seq) converter is a computing process based on RDKit (http://www.rdkit.org) and the characteristics of cyclic peptide sequences. It can extract amino acid units from the cyclic peptide skeleton and match them with the monomer reference library, thereby transforming cyclic peptide SMILES into sequence information. This process mainly relies on the completeness of the monomer reference library. You can access our default monomer reference library on our website. The details of Struc2Seq are available at dfwlab/cyclicpepedia on GitHub.
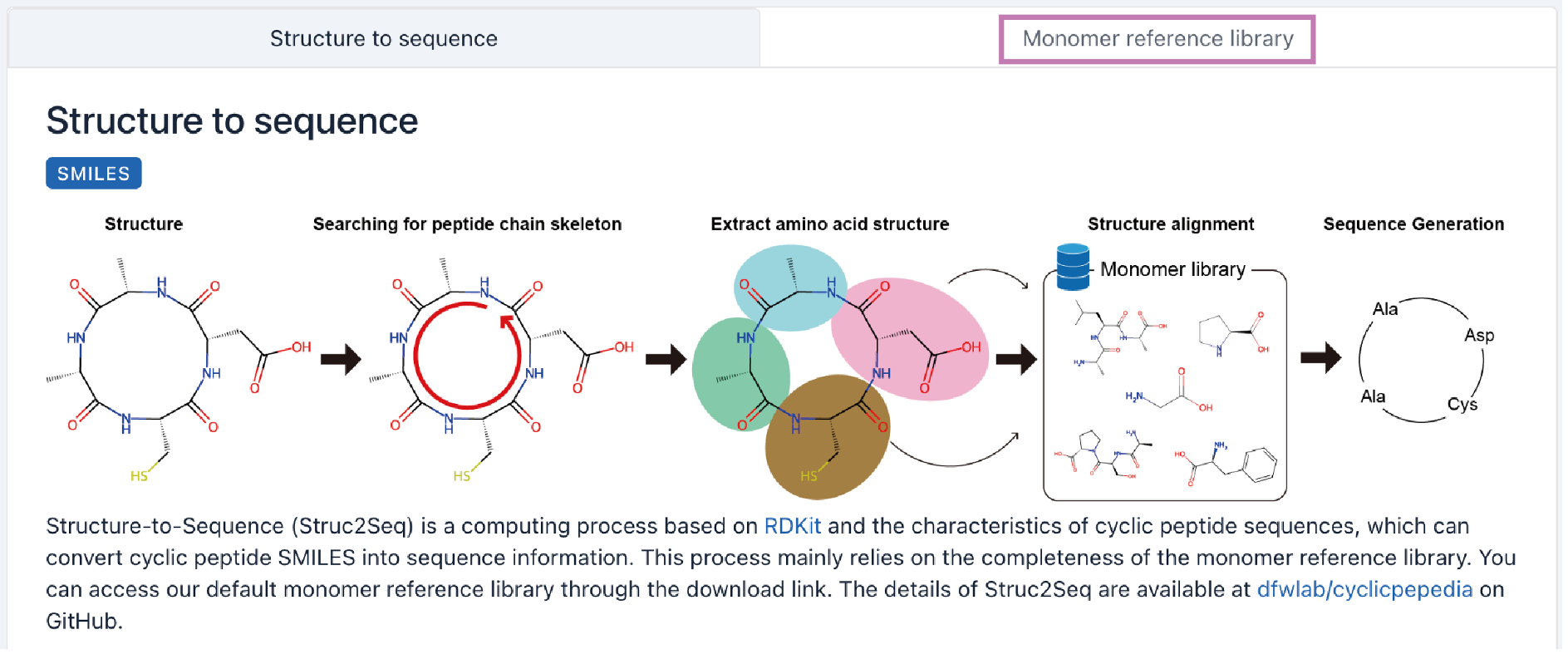
Step 1. Enter your SMILES into the text box.
Example: CC[C@H](C)[C@H]1C(=O)NCC(=O)N[C@H]2C[S@@](=O)C3=C(C[C@@H](C(=O)NCC(=O)N1)NC(=O)[C@@H](NC(=O)[C@@H]4C[C@H](CN4C(=O)[C@@H](NC2=O)CC(=O)N)O)[C@@H](C)[C@H](CO)O)C5=C(N3)C=C(C=C5)O
Step 2. Click the Transform button to perform Struct2Seq conversion.

A detailed Struc2Seq report is provided. It contains the results of each step of Struc2Seq.
a. Check the accuracy of the SMILES.

b. Convert SMILES into atomic structure, identify cyclic peptide skeleton, and renumber atoms.

c. Identify amino acid units.
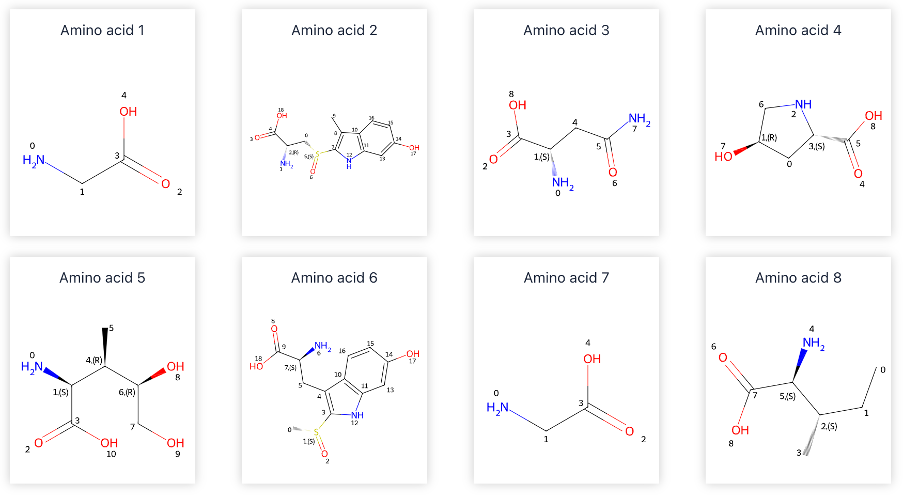
d. Extract individual amino acid units.

e. Map the atomic structures of amino acid units to the monomer reference library.
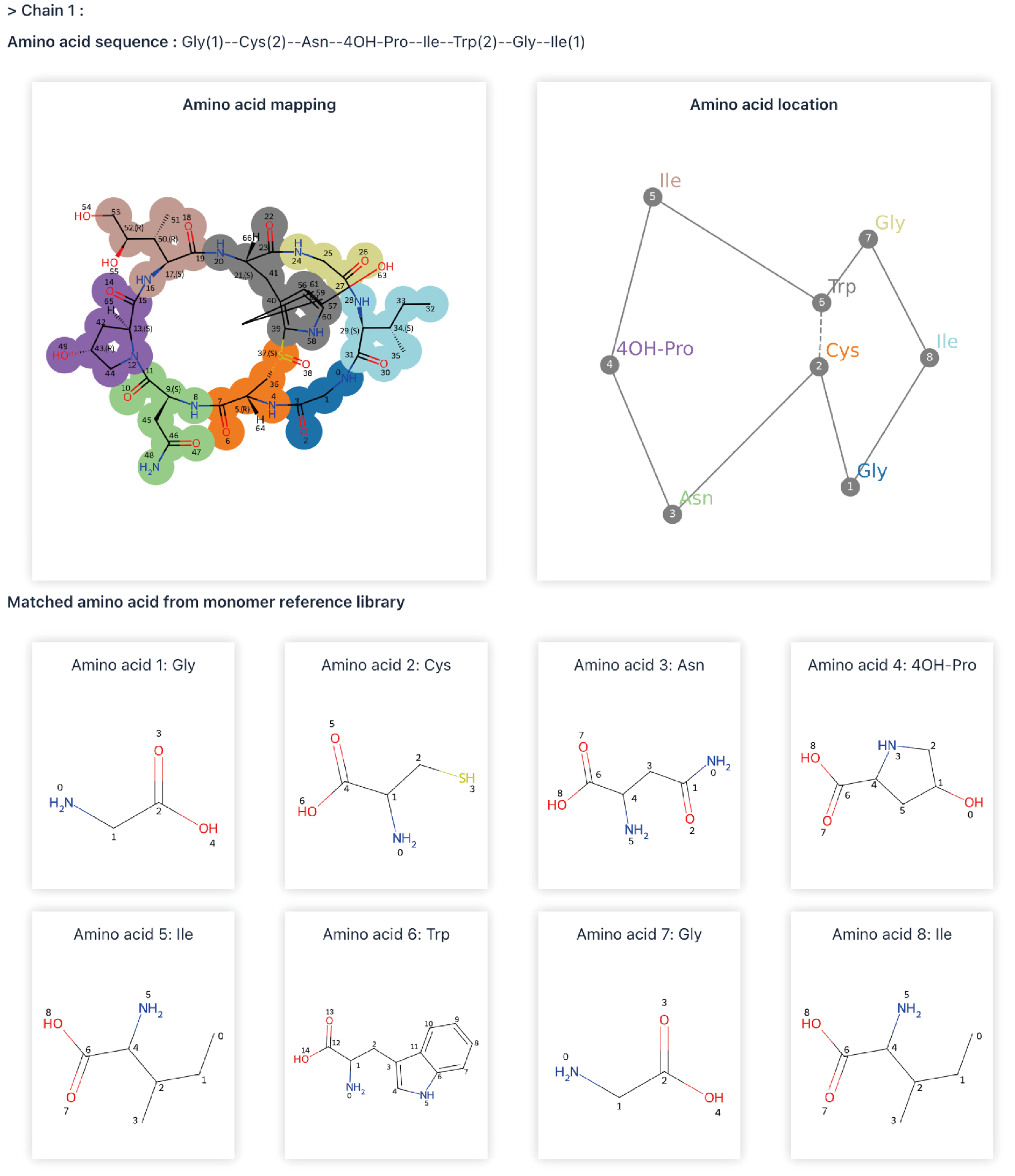
2) Sequence to structure
Click the Tools|Sequence to Structure to enter the Sequence-to-Structure conversion page.
Sequence-to-Structure (Seq2Struc) is a computing process based on RDKit. It can create cyclic peptide sequences and convert sequences to structural information. The details of Seq2Struc are available at dfwlab/cyclicpepedia on GitHub.
Tip! Seq2Struc posits a head-to-tail cyclization, which may not always be the case for all cyclic peptides. Thus, we provide an online editing interface whereby users can refine predicted structures using additional structural information they possess.
i. Seq2Struc for essential amino acids
Step 1. Enter your sequence into the text box. This tool accepts amino acid sequences with one-letter code and three-letter code.
Example:Ala--Ala--Cys--Asp
Step 2. Select the Cyclic parameter.
Step 3. Click the Transform button to perform the Seq2Struc transformation.
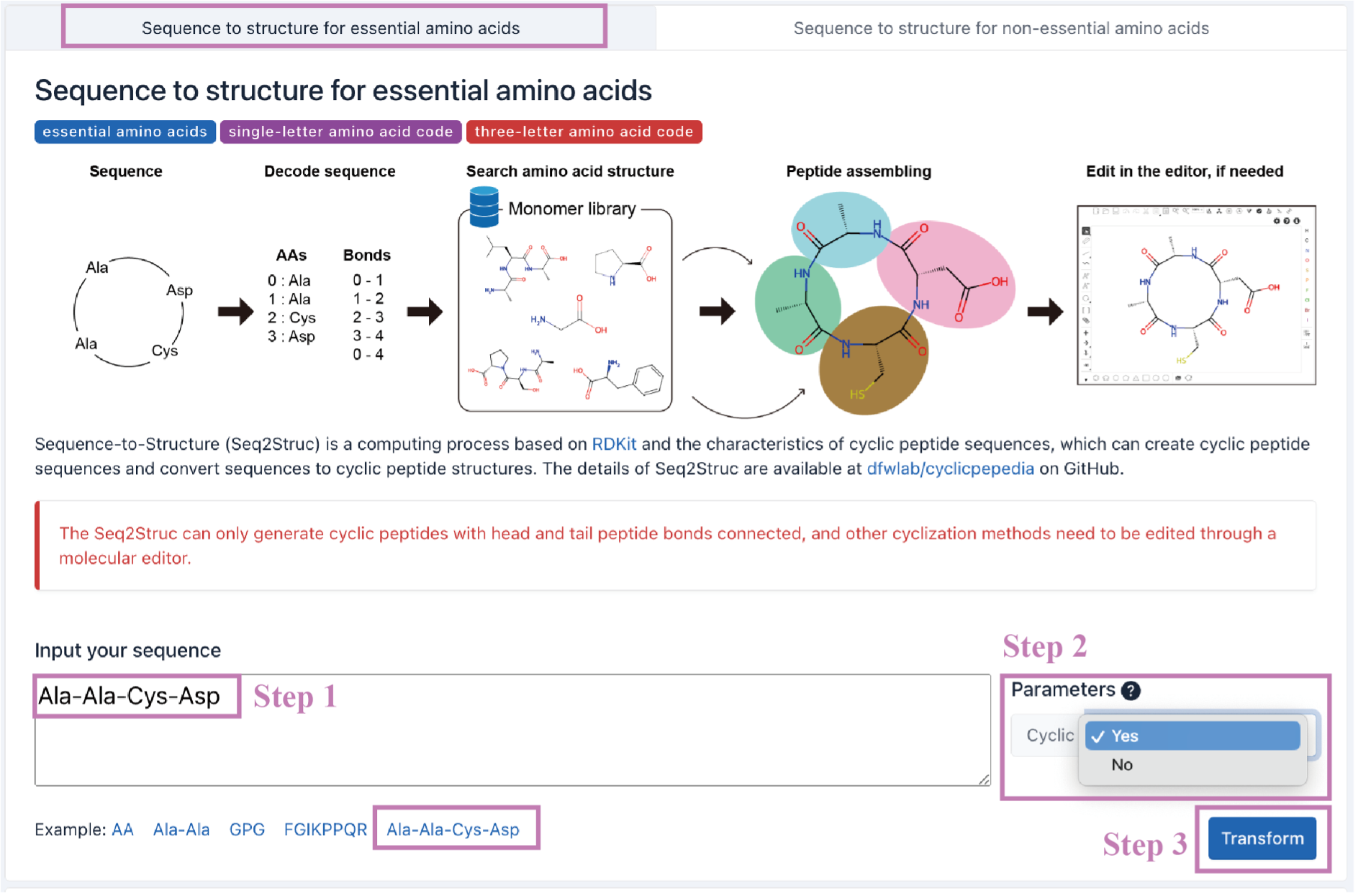
The transformation result will be presented. Click the Open with editor (a) button to enter the online editing interface. The result can be downloaded by clicking the Download report (b) button.
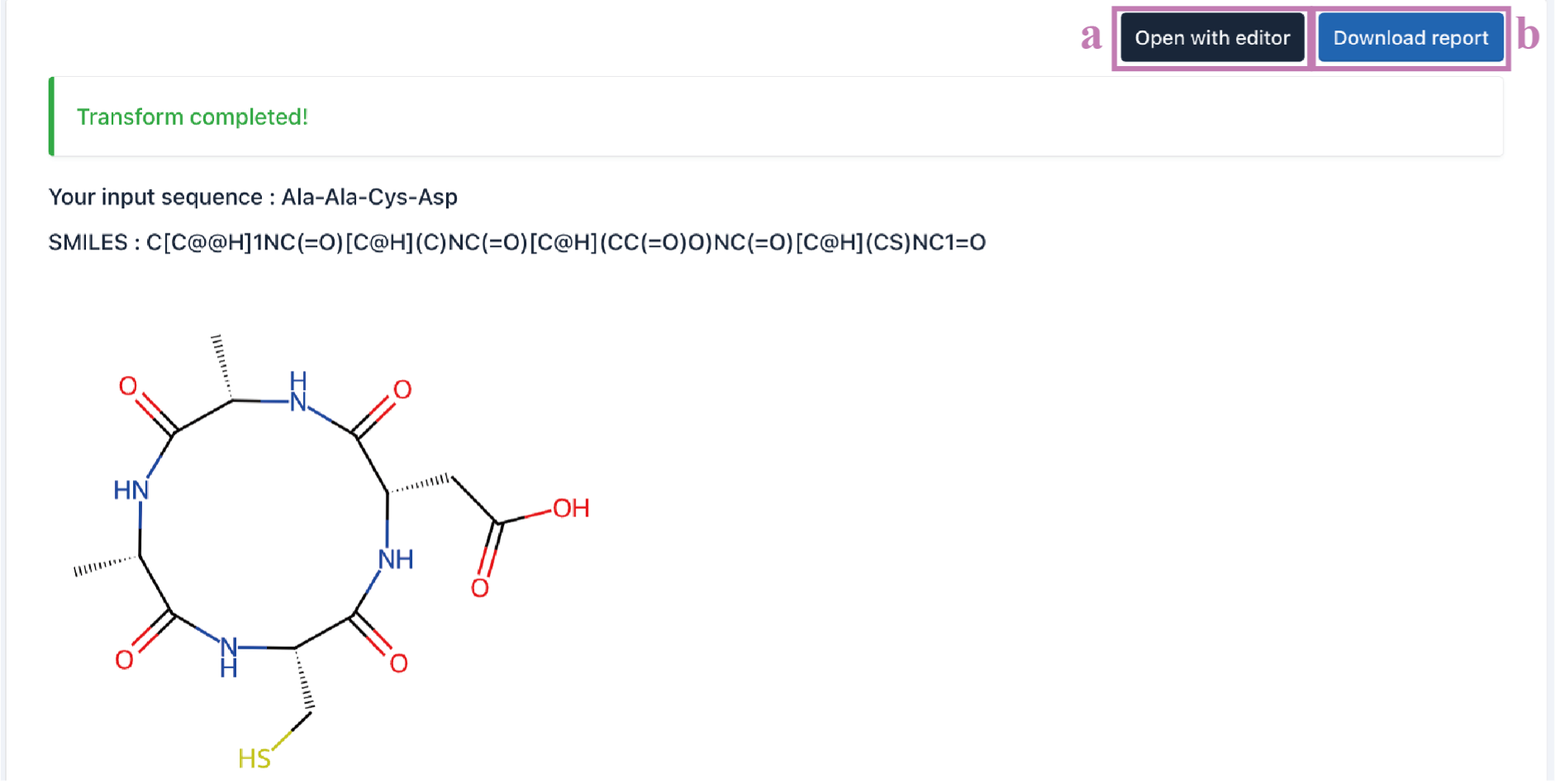
Users can refine the predicted structure by using the online editing interface.
Tip! After editing the structure, remember to click the Save changes (a) button, otherwise it will not be updated in the final result.
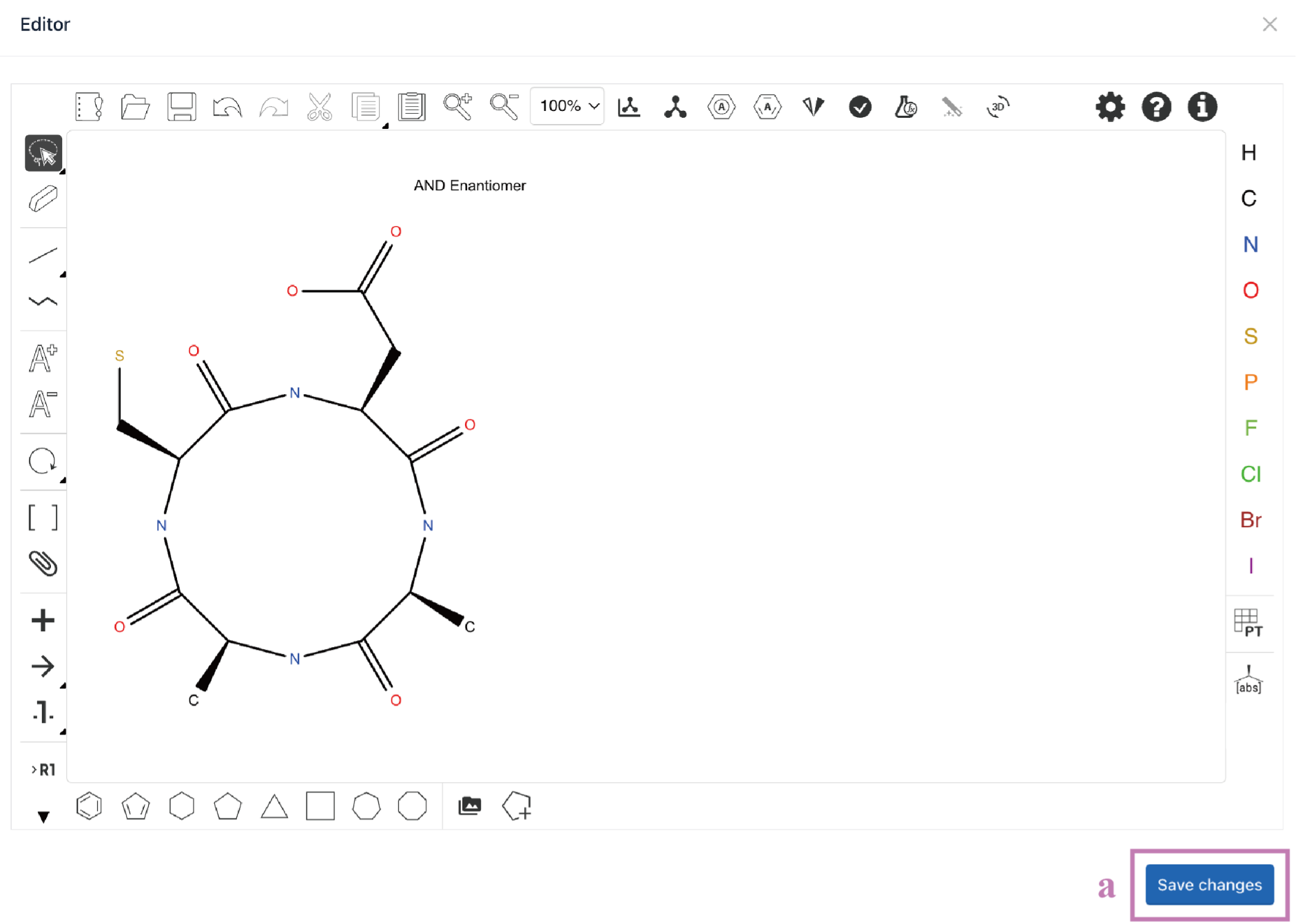
ii. Seq2Struc for non-essential amino acids
The process of Seq2Struc for non-essential amino acids is similar to that of Seq2Struc for essential amino acids. It provides an additional monomer library for users to choose from.
Click the Select from library (a) button to enter the Monomer library.
Over 500 amino acid structural units are provided in our monomer library. Click the monomer name (a) to show its SMILES and atomic structure (b). Add the monomer to your sequence by clicking the Add this monomer to peptide (c) button. The selected monomers are shown in the sequence box (d). Click the Create peptide (e) button to generate a new peptide sequence for Seq2Struc transformation.
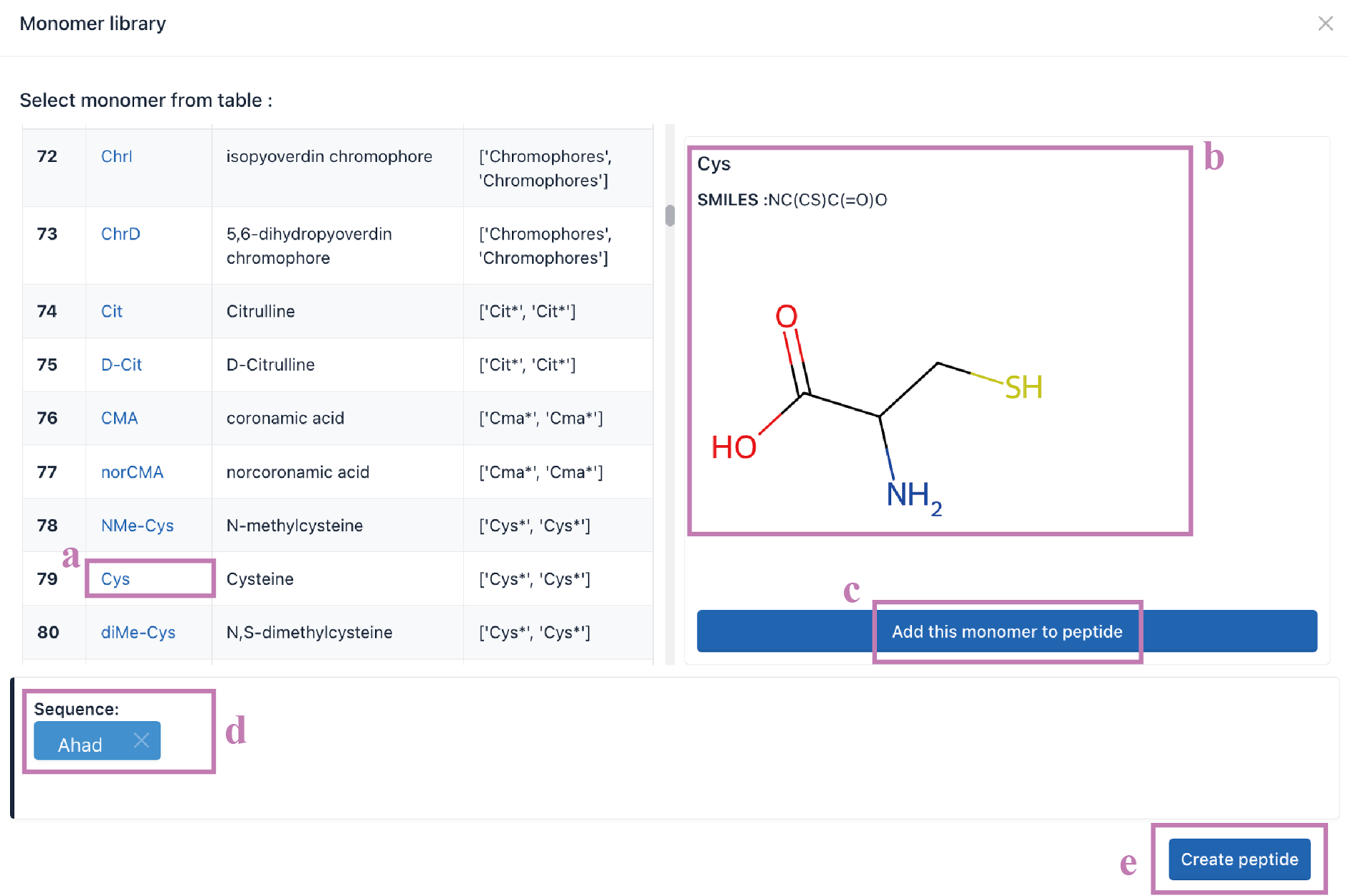
The subsequent steps are the same as Seq2Struc for essential amino acids.
3) Peptide Property Prediction
Click the Tools|Peptide Property Prediction to enter the Peptide Property Prediction page.
i. Chemical and Physical Property Prediction (CPPP)
Chemical and physical properties are computed using RDKit, involving topological polar surface area, complexity, Log(P), hydrogen bond donor count, hydrogen bond acceptor count, rotatable bond count, drug-likeness, and fingerprints. The details of CPPP are available at dfwlab/cyclicpepedia on GitHub.
Step 1. Enter your SMILES into the text box.
Example: CC[C@H](C)[C@H]1C(=O)NCC(=O)N[C@H]2C[S@@](=O)C3=C(C[C@@H](C(=O)NCC(=O)N1)NC(=O)[C@@H](NC(=O)[C@@H]4C[C@H](CN4C(=O)[C@@H](NC2=O)CC(=O)N)O)[C@@H](C)[C@H](CO)O)C5=C(N3)C=C(C=C5)O
Step 2. Click the Predict button to perform CPPP.
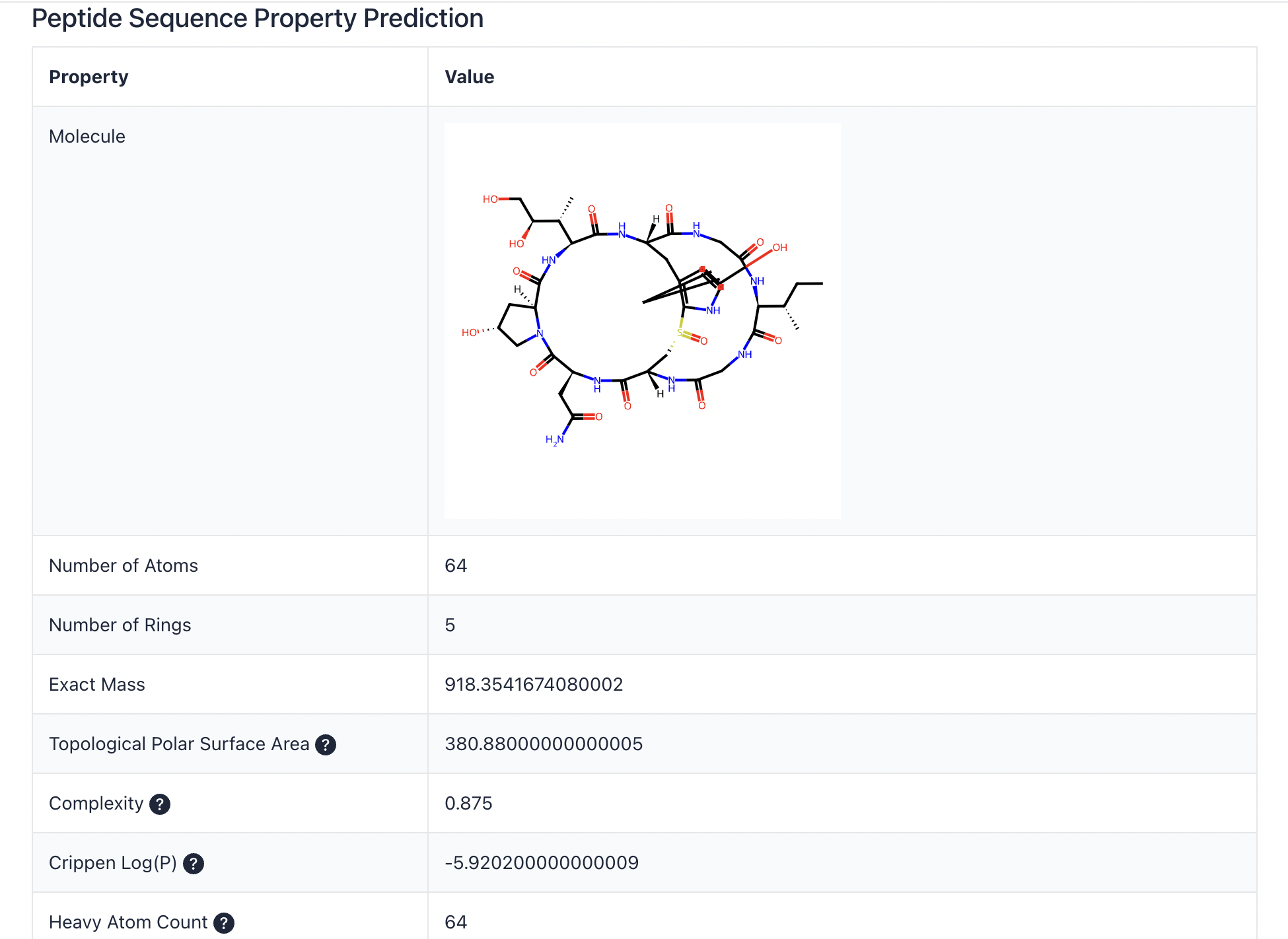
A downloadable report will be provided. Please see Table 1 for a complete list of properties.
Click the Download report button to download the prediction results.
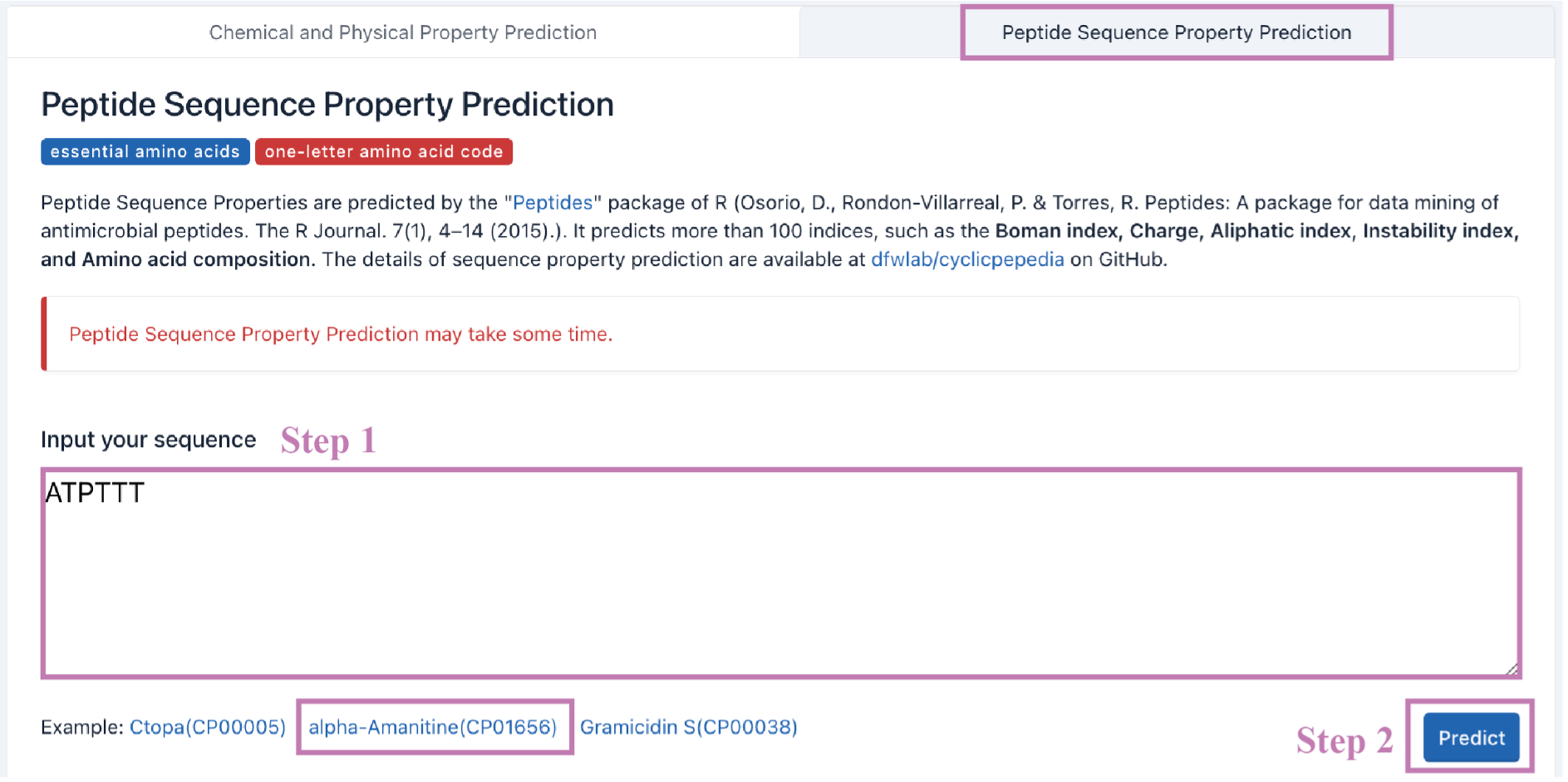
ii. Peptide Sequence Property Prediction (PSPP)
Peptide sequence properties are predicted by the "Peptides" package of R (https://github.com/dosorio/Peptides/). It predicts more than 100 indices, such as the Boman index, charge, aliphatic index, instability index, and amino acid composition. The details of PSPP are available at dfwlab/cyclicpepedia on GitHub.
Step 1. Enter your sequences into the text box. This tool can only be used for peptide sequences in one-letter amino acid code.
Example:ATPTTT
Step 2. Click the Predict button to perform PSPP.
Tip! Peptide Sequence Property Prediction may take some time.
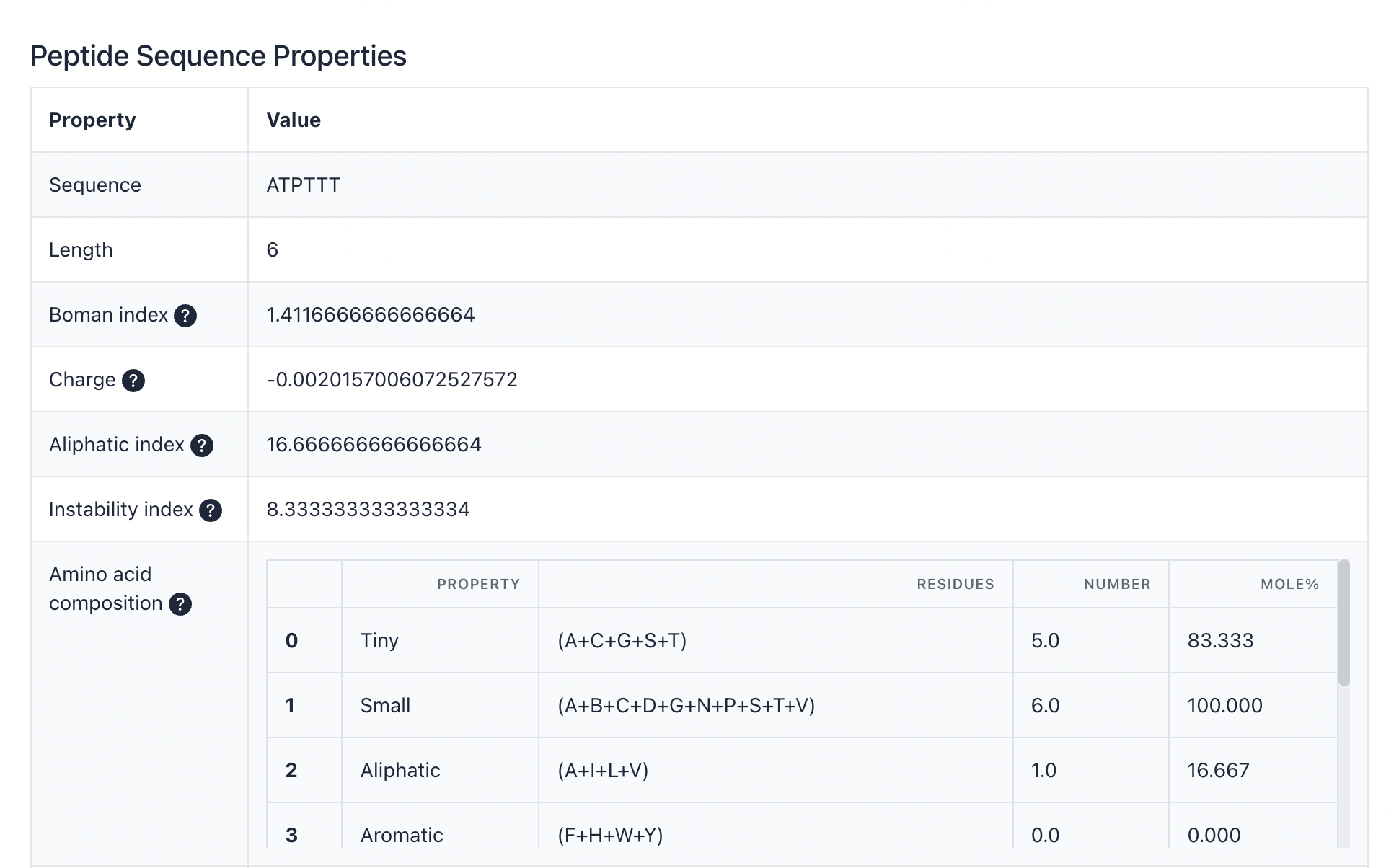
A downloadable report will be provided. Please see Table 2 for a complete list of properties.
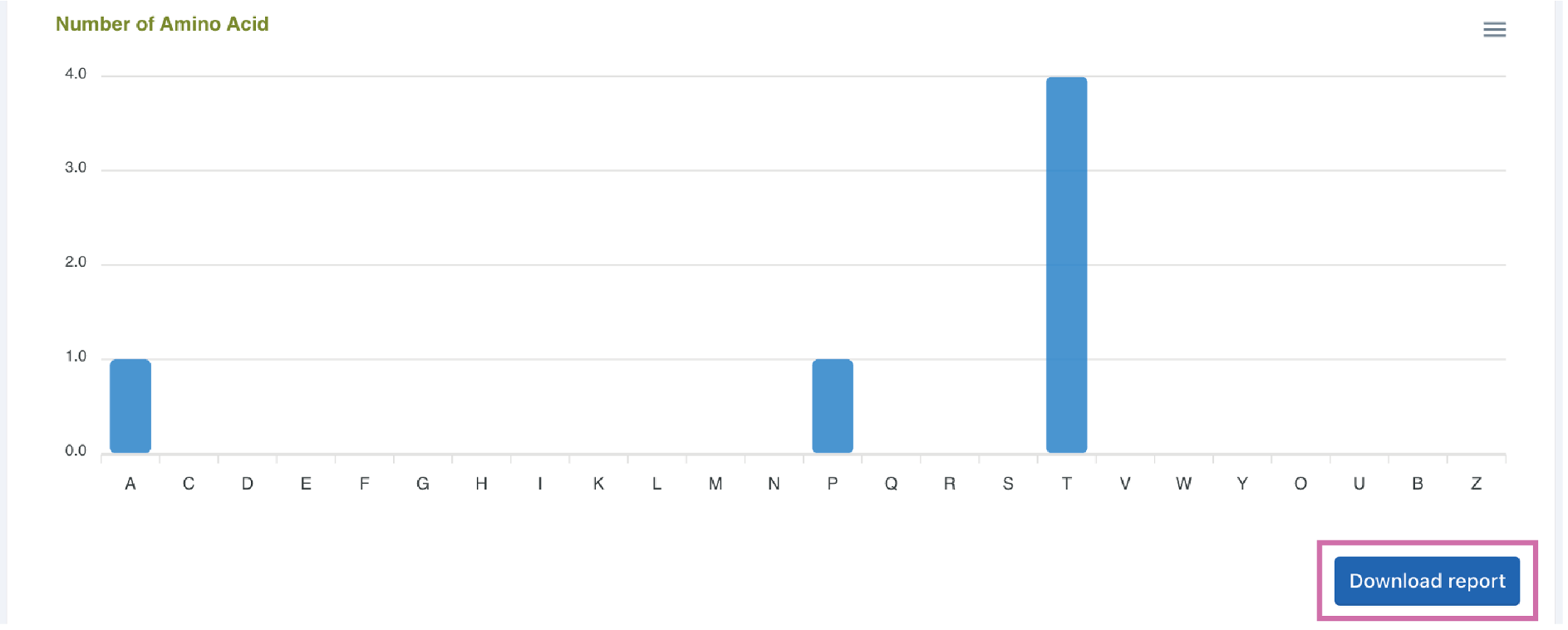
Click the Download report button to download the prediction results.
4) Structure format transformation
Click the Tools|Structure format transformation to enter the Structure format transformation page.
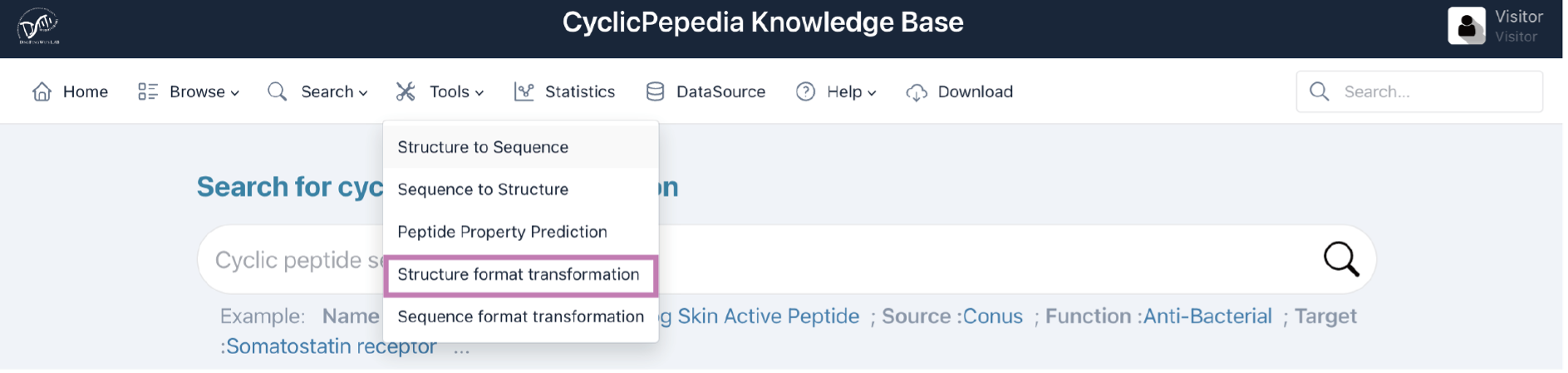
Structure format transformation is a computing process based on RDKit to transform molecules between SMILES, InChI, InChIKey, Mol block, and PDB block formats. The details of this algorithm are available at dfwlab/cyclicpepedia on GitHub.
Step 1. Enter or paste your structural information into the text box. This tool accepts peptide structural data in SMILES, InChI, InChIKey, Mol block, and PDB block formats.
Example:InChI=1S/C50H67N11O11S2/c1-26(62)39(42(53)65)59-49(72)41-50(3,4)74-73-25-38(58-43(66)33(52)21-28-11-6-5-7-12-28)47(70)56-36(22-29-16-18-31(64)19-17-29)45(68)57-37(23-30-24-54-34-14-9-8-13-32(30)34)46(69)55-35(15-10-20-51)44(67)60-40(27(2)63)48(71)61-41/h5-9,11-14,16-19,24,26-27,33,35-41,54,62-64H,10,15,20-23,25,51-52H2,1-4H3,(H2,53,65)(H,55,69)(H,56,70)(H,57,68)(H,58,66)(H,59,72)(H,60,67)(H,61,71)
Step 2. Select the parameters.
3D conformation: Whether to generate a 3D conformation. The 3D conformation is generated by minimizing the energy of Universal Force Field (UFF), which is suitable for small molecule.
Optimize: Whether to perform an optimization.
Tip! It will take some time to create 3D conformations. The generation of PDB block requires additional stereochemical information. The larger the molecule, the longer the optimization time.
Step 3. Click the Transform button to perform the structure format transformation.
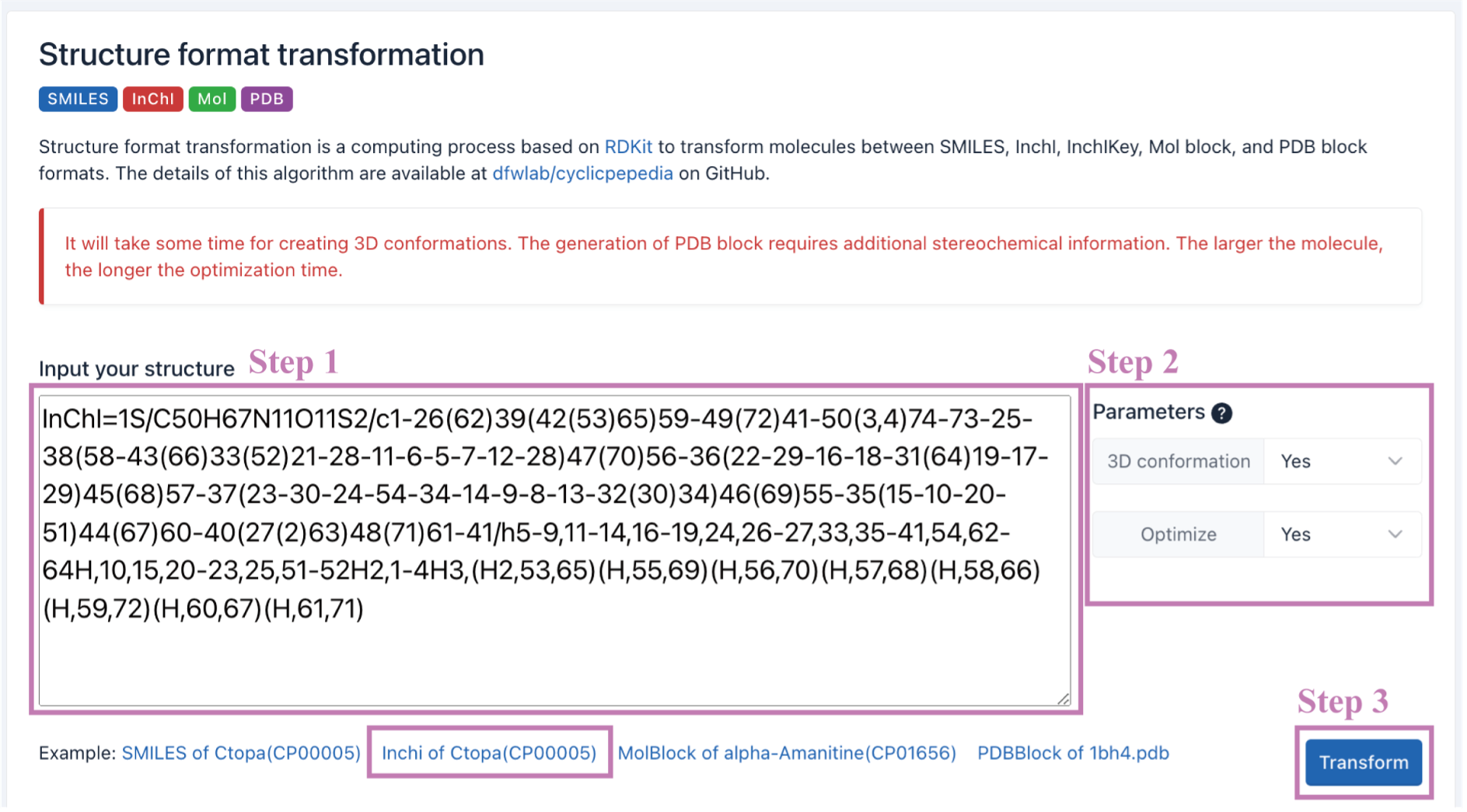
The transformation results will be presented on the webpage. The input structural information will be converted into multiple structure formats, such as SVG, SMILES, InChI, InChIKey, Mol Block, PDB Block, and 3D conformation (if selected). And formats with a download button can be downloaded separately.
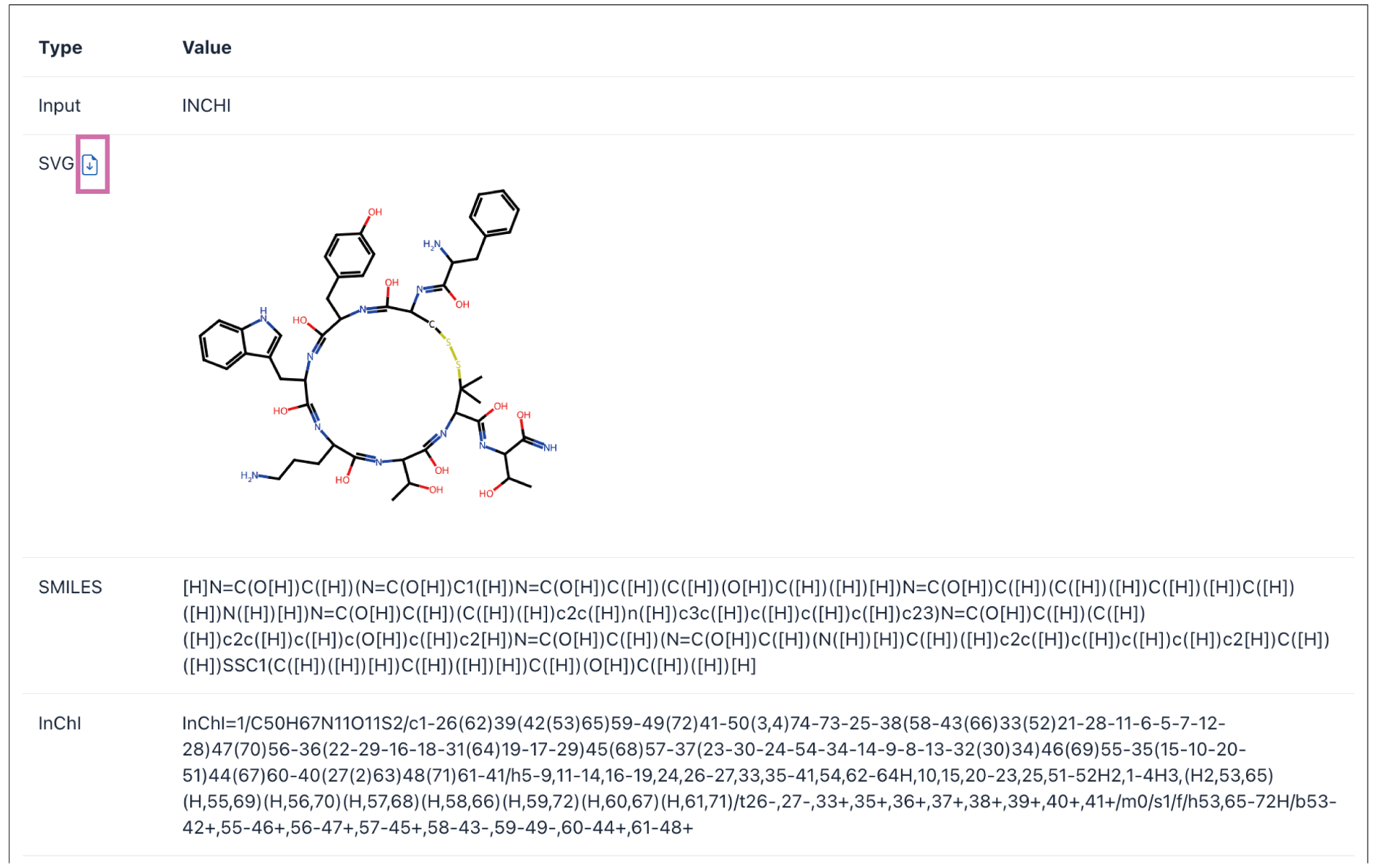
Click the Download report button to download the complete report.
5) Sequence format transformation
Click the Tools|Sequence format transformation to enter the Sequence format transformation page.
Sequence formats such as one-letter code, IUPAC condensed, amino acid chain, graph representation, and sequence graph formats can be inter-converted through the Sequence format transformation tool. A description of these formats is available online (a) and in Table 3. The details of this algorithm are available at dfwlab/cyclicpepedia on GitHub.
Step 1. Paste or enter your sequence in one-letter code, IUPAC condensed, amino acid chain, graph representation, or sequence graph formats.
Example: cyclo[DL-Cys(1)-DL-Cys-DL-Val(1)-DL-Leu-DL-xiIle]
Step 2. Click the Transform button to perform the Sequence format transformation.
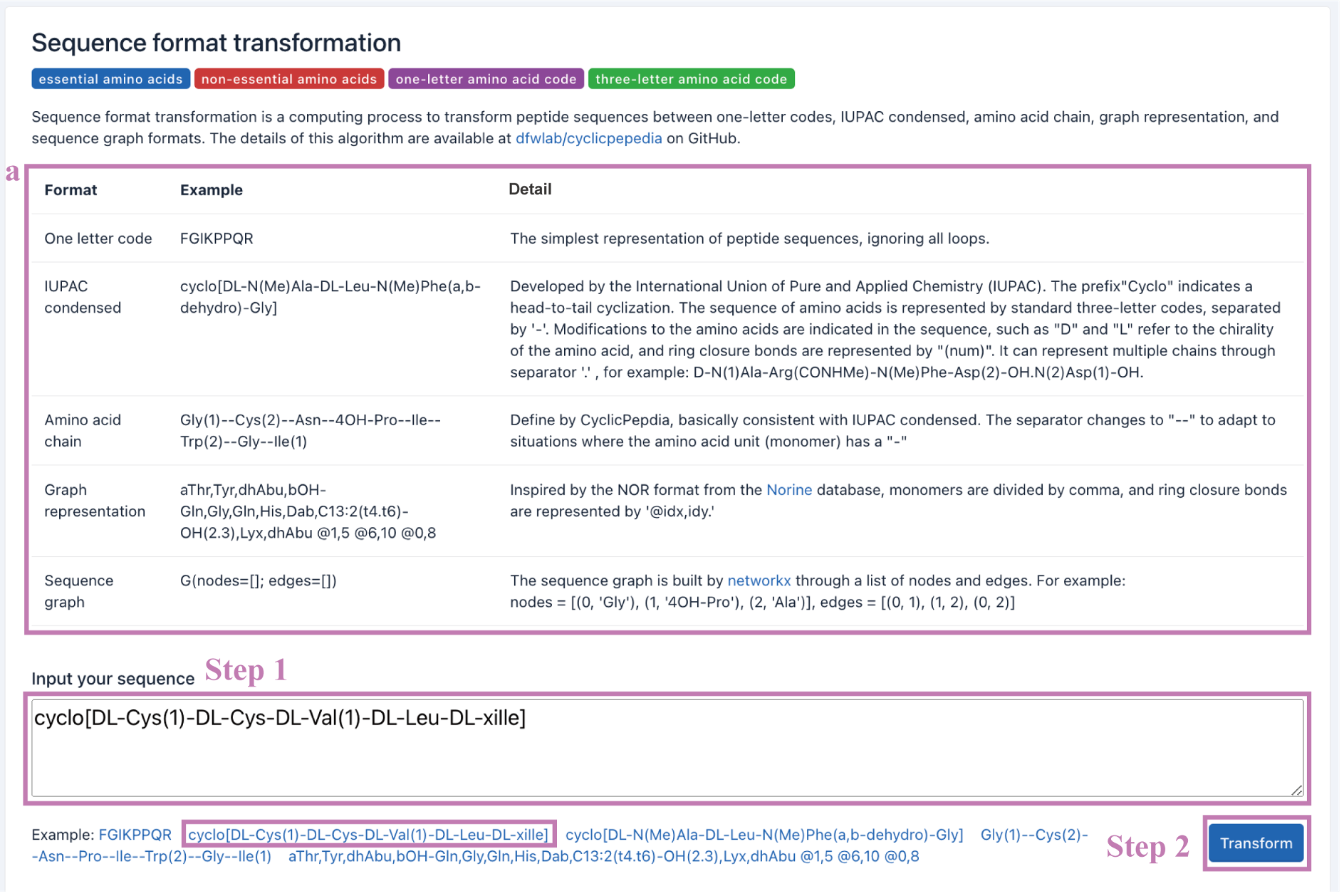
The transformation results will be presented on the webpage. The input sequence information will be converted into multiple sequence formats. And formats with a download button can be downloaded separately.

Table 1. List of the chemical and physical properties.
| Property | Description |
|---|---|
| Number of Atoms | |
| Number of Rings | |
| Exact Mass | |
| Topological Polar Surface Area | Measures the surface area occupied by polar atoms, often used to predict drug transport properties. |
| Complexity | Indicates the structural complexity of the molecule, with higher values representing more intricate structures. |
| Crippen Log(P) | Represents the logarithm of the partition coefficient between n-octanol and water, used to estimate the molecule's hydrophobicity. |
| Heavy Atom Count | Counts the number of non-hydrogen atoms in the molecule, reflecting its size and complexity. |
| Hydrogen Bond Donor Count | The number of atoms in the molecule that can donate hydrogen bonds, important for molecular interactions. |
| Hydrogen Bond Acceptor Count | The number of atoms capable of accepting hydrogen bonds, crucial for molecular recognition and binding. |
| Rotatable Bond Count | Counts the number of bonds that allow free rotation around themselves, affecting the molecule's flexibility. |
| Formal Charge | The overall electric charge of the molecule, with zero indicating a neutral molecule. |
| Refractivity | Measures the molecule's ability to refract light, related to polarizability and electronic properties. |
| Rule of Five | Indicates non-compliance with Lipinski's rule of five, suggesting potential issues with bioavailability as an oral drug. |
| Veber's Rule | Shows non-adherence to Veber's rules, potentially impacting oral bioavailability and permeability. |
| Ghose Filter | A molecular property filter used to assess the drug-likeness of a compound based on its physicochemical properties. |
| RDKit Fingerprint | This is an RDKit-specific fingerprint. |
| Daylight-like Fingerprint | This is an RDKit-specific fingerprint that is inspired by (though it differs significantly from) public descriptions of the Daylight fingerprint. |
| Morgan Fingerprint | The RDKit implementation uses the feature types Donor, Acceptor, Aromatic, Halogen, Basic, and Acidic. |
| MACCS Keys | SMARTS definitions for the publicly available MACCS keys and a MACCS fingerprinter. |
Table 2. List of the peptide sequence properties.
| Property | Description |
|---|---|
| Sequence | |
| Length | Sequence length |
| Boman index | This property computes the potential protein interaction index proposed by Boman (2003) based in the amino acid sequence of a protein. The index is equal to the sum of the solubility values for all residues in a sequence, it might give an overall estimate of the potential of a peptide to bind to membranes or other proteins as receptors, to normalize it is divided by the number of residues. A protein have high binding potential if the index value is higher than 2.48. |
| Charge | This property computes the net charge of a protein sequence based on the Henderson-Hasselbalch equation described by Moore, D. S. (1985). The net charge can be calculated at defined pH using one of the 9 pKa scales availables: Bjellqvist, Dawson, EMBOSS, Lehninger, Murray, Rodwell, Sillero, Solomon or Stryer. |
| Aliphatic index | This property calculates the Ikai (1980) aliphatic index of a protein. The aindex is defined as the relative volume occupied by aliphatic side chains (Alanine, Valine, Isoleucine, and Leucine). It may be regarded as a positive factor for the increase of thermostability of globular proteins. |
| Instability index | This property calculates the instability index proposed by Guruprasad (1990). This index predicts the stability of a protein based on its amino acid composition, a protein whose instability index is smaller than 40 is predicted as stable, a value above 40 predicts that the protein may be unstable. |
| Amino acid composition | Tiny, Small, Aliphatic, Aromatic, Non-polar, Polar, Charged, Basic and Acidic based on their size and R-groups using same function implemented in EMBOSS 'pepstat'. |
| BLOSUM62 | BLOSUM indices were derived of physicochemical properties that have been subjected to a VARIMAX analyses and an alignment matrix of the 20 natural AAs using the BLOSUM62 matrix. |
| Cruciani properties | The Cruciani properties of an amino-acids sequence is calculated using the scaled principal component scores that summarize a broad set of descriptors calculated based on the interaction of each amino acid residue with several chemical groups (or 'probes'), such as charged ions, methyl, hydroxyl groups, and so forth. |
| FASGAI vectors | The FASGAI vectors (Factor Analysis Scales of Generalized Amino Acid Information) is a set of amino acid descriptors, that reflects hydrophobicity, alpha and turn propensities, bulky properties, compositional characteristics, local flexibility, and electronic properties, that can be utilized to represent the sequence structural features of peptides or protein motifs. |
| Hydrophobic moment | This properties compute the hmoment based on Eisenberg, D., Weiss, R. M., & Terwilliger, T. C. (1984). Hydriphobic moment is a quantitative measure of the amphiphilicity perpendicular to the axis of any periodic peptide structure, such as the a-helix or b-sheet. It can be calculated for an amino acid sequence of N residues and their associated hydrophobicities Hn. |
| Hydrophobicity index | This property calculates the GRAVY hydrophobicity index of an amino acids sequence using one of the 38 scales from different sources. |
| Kidera factors | The Kidera Factors were originally derived by applying multivariate analysis to 188 physical properties of the 20 amino acids and using dimension reduction techniques. This function calculates the average of the ten Kidera factors for a protein sequence. |
| Theoretical class | This property calculates the theoretical class of a protein sequence based on the relationship between the hydrophobic moment and hydrophobicity scale proposed by Eisenberg (1984). |
| MS-WHIM scores | MS-WHIM scores were derived from 36 electrostatic potential properties derived from the three-dimensional structure of the 20 natural amino acids. |
| Isoelectic point (pI) | The isoelectric point (pI), is the pH at which a particular molecule or surface carries no net electrical charge. |
| protFP | The ProtFP descriptor set was constructed from a large initial selection of indices obtained from the AAindex database for all 20 naturally occurring amino acids. |
| ST-scales | ST-scales were proposed by Yang et al, taking 827 properties into account which are mainly constitutional, topological, geometrical, hydrophobic, elec- tronic, and steric properties of a total set of 167 AAs. |
| T-scales | T-scales are based on 67 common topological descriptors of 135 amino acids. These topological descriptors are based on the connectivity table of amino acids alone, and to not explicitly consider 3D properties of each structure. |
| VHSE-scales | VHSE-scales (principal components score Vectors of Hydrophobic, Steric, and Electronic properties), is derived from principal components analysis (PCA) on independent families of 18 hydrophobic properties, 17 steric properties, and 15 electronic properties, respectively, which are included in total 50 physicochemical variables of 20 coded amino acids. |
| Z-scales | Z-scales are based on physicochemical properties of the AAs including NMR data and thin-layer chromatography (TLC) data. |
| Amino acid count |
Table 3. Sequence formats.
| Format | Example | Description |
|---|---|---|
| One letter code | FGIKPPQR | The simplest representation of peptide sequences, ignoring all loops. |
| IUPAC condensed | cyclo[DL-N(Me)Ala-DL-Leu-N(Me)Phe(a,b-dehydro)-Gly] | Developed by the International Union of Pure and Applied Chemistry (IUPAC). The prefix"Cyclo" indicates a head-to-tail cyclization. The sequence of amino acids is represented by standard three-letter codes, separated by '-'. Modifications to the amino acids are indicated in the sequence, such as "D" and "L" refer to the chirality of the amino acid, and ring closure bonds are represented by "(num)". It can represent multiple chains through separator '.' , for example: D-N(1)Ala-Arg(CONHMe)-N(Me)Phe-Asp(2)-OH.N(2)Asp(1)-OH. |
| Amino acid chain | Gly(1)--Cys(2)--Asn--4OH-Pro--Ile--Trp(2)--Gly--Ile(1) | Define by CyclicPepdia, basically consistent with IUPAC condensed. The separator changes to "--" to adapt to situations where the amino acid unit (monomer) has a "-" |
| Graph representation | aThr,Tyr,dhAbu,bOH-Gln,Gly,Gln,His,Dab,C13:2(t4.t6)-OH(2.3),Lyx,dhAbu @1,5 @6,10 @0,8 | Inspired by the NOR format from the Norine |
| database, monomers are divided by comma, and ring closure bonds are represented by '@idx,idy.' | ||
| Sequence graph | G(nodes=[]; edges=[]) | The sequence graph is built by Networkx |
| through a list of nodes and edges. For example:nodes = [(0, 'Gly'), (1, '4OH-Pro'), (2, 'Ala')], edges = [(0, 1), (1, 2), (0, 2)] |