
CyclicPepedia Knowledge Base
Local alignment (Smith-Waterman)
essential amino acids single-letter amino acid code linear peptide
The Smith-Waterman algorithm is utilized for the local alignment of peptide sequences (Biopython: pairwise2.align.localxx). This method allows for parameter adjustment to set penalties for matches, mismatches, and gaps, ensuring tailored alignment based on specific sequence characteristics. It should be noted that this method is only applicable to peptides represented by single-letter amino acid codes.
Graph alignment
all amino acids three-letter amino acid code cyclic peptide
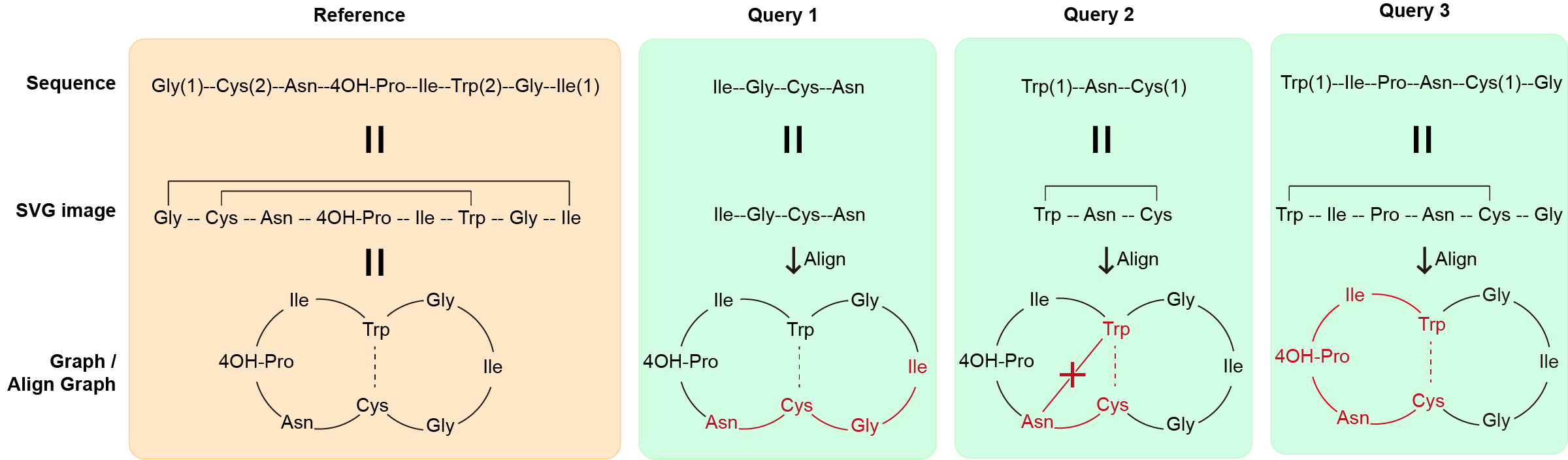
The graph alignment is a custom algorithm developed by CyclicPepedia, specifically designed for cyclic peptide sequence alignment. The alignment algorithm based on networkx extension can convert cyclic peptides into graphical structures and measure the similarity by Graph Isomorphism. The above is a schematic diagram of the algorithm. The input sequence format can be IUPAC condensed, Amino acid chain and Graph presentation (refer to Sequence format transform for sequence examples).
It may take some time for long peptide sequences. You can adjust parameters to reduce search space (e.g., set min AA <= sequence length <= max AA and set a higher value for AA comp threshold).