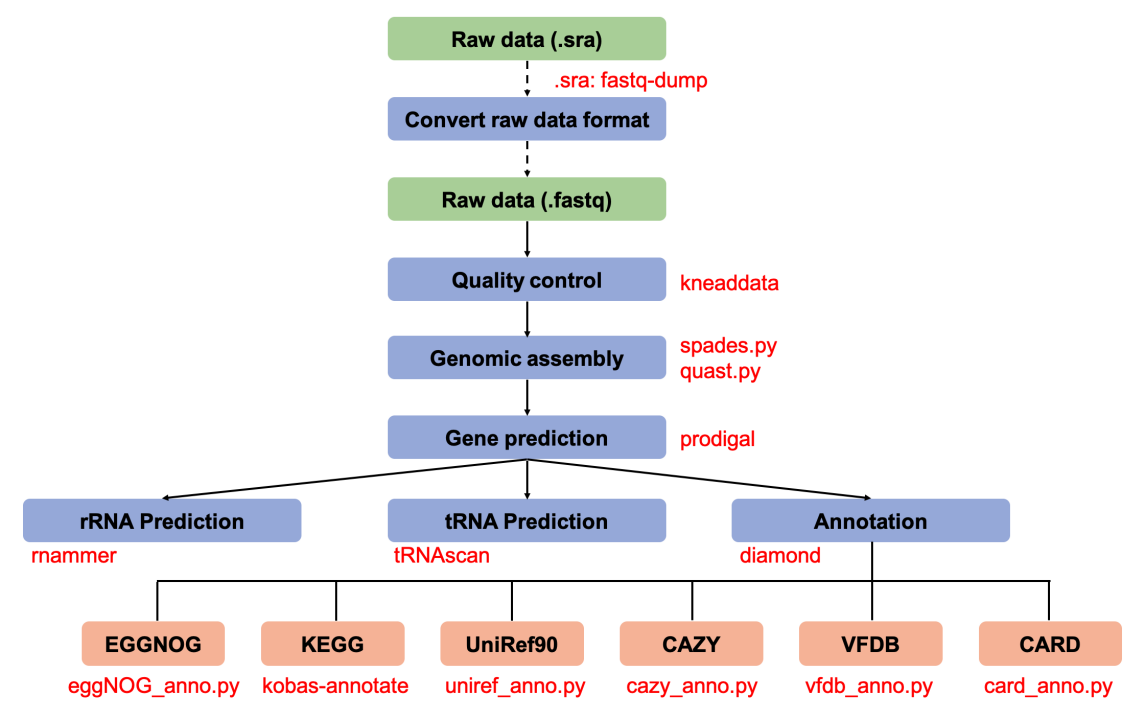
The genome-analysis pipeline is used to microbial genome annotation. For each sample, this pipeline includes the steps of quality control (KneadData), metagenomic assembly (Spades), gene prediction (Prodigal or MetaGeneMark). The assembly results are run by RNAmmer to predict the location of rRNAs and analyzed by tRNAscan-SE to search for tRNA genes in genomic sequences. Finally, the amino acid sequences of the gene set are aligned (DIAMOND blastp) with the sequences of six common functional databases including EGGNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups), KEGG (Kyoto Encyclopedia of Genes and Genomes), UniRef90 (The UniProt Reference Clusters 90), CAZY (Carbohydrate-Active enZYmes Database), VFDB (Virulence Factor Database) and CARD (The Comprehensive Antibiotic Resistance Database), thus obtain the functional abundance profile for each sample.
The steps of genome-assembly pipeline (for more information, please refer to user manual)
1、Register an account. If you don’t have an iMAC account, please click here to register your account.
2、Login. If you already have an iMAC account, please click login in the navigation bar.
3、Upload data. Download the FTP client software FileZilla, and log in sftp://fms.biosino.org:44399 using the registered user name and password. Then, upload your data through FileZilla and browse them in user center/My Data module.
4、Create new task. In the user center/Task List/genome-analysis Task, click “New Task” to create a genome-analysis pipeline task. Then, adjust the default parameters according to your requirement and submit.
5、Download results. After the task is finished, you can download the results in the user center/Task List/genome-analysis Task and click the eye icon to view interactive graphics.