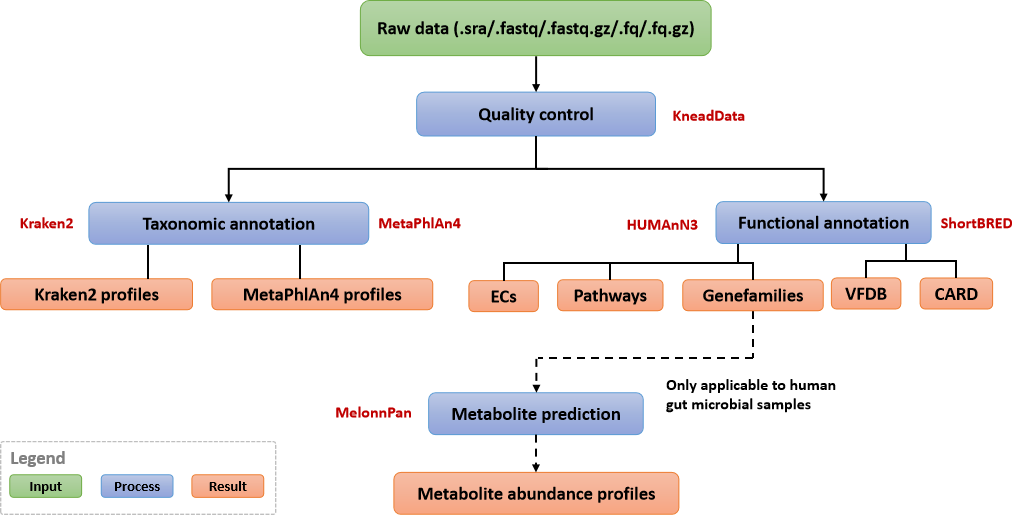
The mWGS-reads pipeline is based on a reads mapping method, and mainly includes the steps of quality control (KneadData), construction of taxonomic abundance profile (MetaPhlAn4, Kraken2), construction of functional abundance profile (HUMAnN3) and functional annotation (ShortBRED) for VFDB (Virulence Factor Database) and CARD (The Comprehensive Antibiotic Resistance Database) databases. For gut microbial samples, the metabolites abundance profile can also be obtained by Melonnpan. The current pipeline supports the removal of human and mouse host contamination.
The steps of mWGS-reads pipeline (for more information, please refer to user manual)
1、Register an account. If you don’t have an iMAC account, please click here to register your account.
2、Login. If you already have an iMAC account, please click login in the navigation bar.
3、Upload data. Download the FTP client software FileZilla, and log in sftp://fms.biosino.org:44399 using the registered user name and password. Then, upload your data through FileZilla and browse them in user center/My Data module.
4、Create new task. In the user center/Task List/mWGS-reads Task, click “New Task” to create a mWGS-reads pipeline task. Then, adjust the default parameters according to your requirement and submit.
5、Download results. After the task is finished, you can download the results in the user center/Task List/mWGS-reads Task and click the eye icon to view interactive graphics.