 Related
RelatedAlphaFold2-PointSite-Docking
AlphaFold2-PointSite-Docking is a pipline for the 3D structure prediction, accurate identification of protein ligand binding atoms and prediction of the ligand-receptor complex structure of catalytic bioparts.

PVQD
PVQD is a method based on deep learning for protein structure design and prediction. Here we published the source code and the demos for PVQD. This code is developed on Torch framework.
PathFinder
PathFinder is a tool for the identification of pathways basing on the experimental verified reactions for catalytic bioparts.
BiopartFinder
BiopartFinder is a tool for the general catalytic biopart sequence identification and similarity searches.
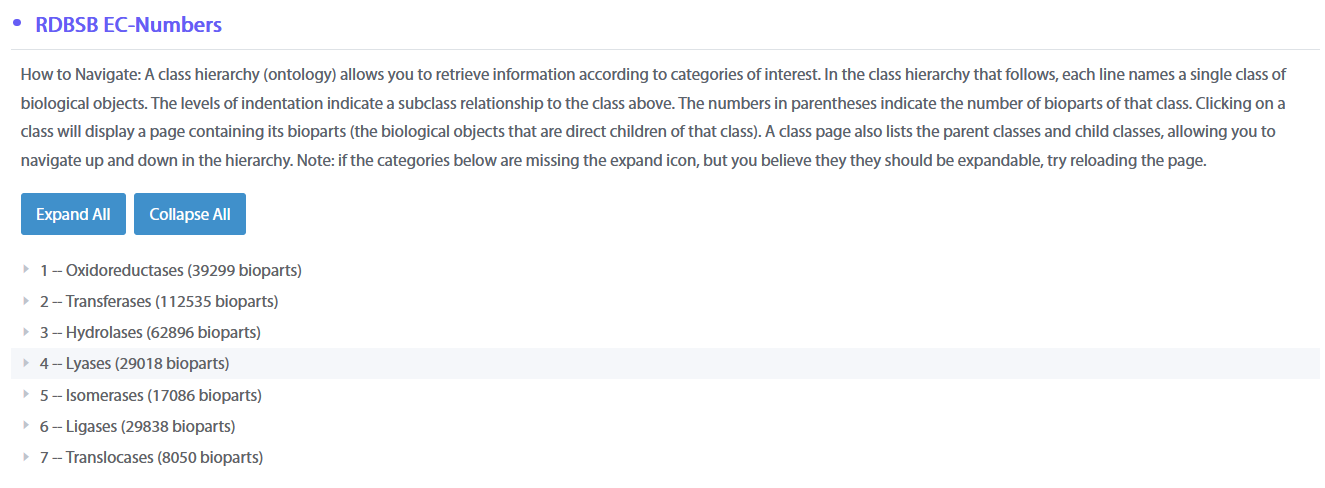
RDBSB EC-Numbers
A class hierarchy (ontology) allows you to retrieve information according to categories of interest. In the class hierarchy that follows, each line names a single class of biological objects. The levels of indentation indicate a subclass relationship to the class above. The numbers in parentheses indicate the number of bioparts of that class. Clicking on a class will display a page containing its bioparts (the biological objects that are direct children of that class). A class page also lists the parent classes and child classes, allowing you to navigate up and down in the hierarchy. Note: if the categories below are missing the expand icon, but you believe they they should be expandable, try reloading the page.
GTDB
GTDB is an integrated repository of glycosyltransferases, which collects comprehensive information, including amino acid sequences, coding region sequences, available tertiary structures, protein classification families, catalytic reactions and metabolic pathways involved, from distinct well-known databases or predictions.
RefMetaDB
Reference Metabolome Database for Plants (RefMetaPlant) serves as an integrated database and analysis platform dedicated to becoming the centralized resource for plant metabolomic research. It aims to standardize and integrate the reference metabolome data, providing a comprehensive platform for researchers in plant metabolomics, genetics, and related fields. Currently, RefMetaPlant 1.0 is released to provided:
(1) 1,086,000+ experimental mass spectra we obtained using UPLC coupled with Quadrupole-Orbitrap High Resolution Mass Spectrometer (UPLC-Q-Orbitrap-HRMS) on samples of 150+ plant species from Bryophyta, Lycopodiopsida, Pteridophyta, Gymnospermae, and Angiospermae;
(2) The reference metabolome for 153 plant species across the five major phyla of green plants;
(3) 325,100+ standard compounds mass spectral data in a library, which include data of 135,464 experimental reference mass spectral from public databases like MassBank, MoNA, Respect, FiehnLib, RIKEN PlaSMA, and data of 189,639 in silico mass spectra;
(4) A set of related query and analytical tools like ‘LC-MS/MS Query’, 'RefMetaBlast' and 'CompoundLibBlast' for plants metabolome search and profiling, and metabolite identification.
RefMetaPlant provides a powerful platform to support plant genome-scale metabolomics analysis, and promote knowledge/data sharing and collaborations of metabolomic research.
tmap
For large scale and integrative microbiome research, it is expected to apply advanced data mining techniques in microbiome data analysis.
Topological data analysis (TDA) provides a promising technique for analyzing large scale complex data. The most popular Mapper algorithm is effective in distilling data-shape from high dimensional data, and provides a compressive network representation for pattern discovery and statistical analysis.
tmap is a topological data analysis framework implementing the TDA Mapper algorithm for population-scale microbiome data analysis. We developed tmap to enable easy adoption of TDA in microbiome data analysis pipeline, providing network-based statistical methods for enterotype analysis, driver species identification, and microbiome-wide association analysis of host meta-data.
ABACUS2
The ABACUS (a backbone-based amino acid usage survey) method uses unique statistical energy functions to carry out protein sequence design. Although some of its results have been experimentally verified, its accuracy remains improvable because several important components of the method have not been specifically optimized for sequence design or in contexts of other parts of the method. The computational efficiency also needs to be improved to support interactive online applications or the consideration of a large number of alternative backbone structures.
We derived a model to measure solvent accessibility with larger mutual information with residue types than previous models, optimized a set of rotamers which can approximate the sidechain atomic positions more accurately, and devised an empirical function to treat inter-atomic packing with parameters fitted to native structures and optimized in consistence with the rotamer set. Energy calculations have been accelerated by interpolation between pre-determined representative points in high-dimensional structural feature spaces. Sidechain repacking tests showed that ABACUS2 can accurately reproduce the conformation of native sidechains. In sequence design tests, the native residue type recovery rate reached 37.7%, exceeding the value of 32.7% for ABACUS1. Applying ABACUS2 to designed sequences on three native backbones produced proteins shown to be well-folded by experiments.
TFBS
TFBS, Binding Sites Prediction of TetR Family Repressors
This server can be used to predict DNA binding sites for transcription factors of TetR Family Repressors (TFRs). Two methods as described in the reference at the end of this page are used: a genome sequence-based method which uses ideas of phylogenetic footprinting, and a statistical energy-based method which calculates sequence energies of DNA octamers given the amino acid sequences of TFRs. Without user-provided DNA sequences, TFBSs are predicted as genome sequence fragments near the ORF of a query TFR, and as octamer DNA sequences and sequence logos representing predicted half binding sites of a query TFR. If a DNA sequence is provided by user, the given DNA sequence will be scanned for most possible TFBSs.
ABACUS-R
ABACUS-R is a method based on deep learning for designing amino acid sequences that autonomously fold into a given target backbone.
SCUBA
SCUBA (SideChain Unspecialized Backbone Arrangement) is a statistical energy function of protein conformation. It consists of energy terms derived from known protein structures using a novel adaptive-kernel neighbor counting-neural network(NC-NN) approach. It is continuous with analytical gradients, allowing protein structures to be sampled and/or optimized with complete flexibility by stochastics dynamics (SD) simulations.
By design, SCUBA energy contains both local and through-space packing interactions of mainchain atoms, while sidechains in the model mainly serve as steric placeholders. In SCUBA-driven protein backbone design, SD simulated annealing can be applied to generate optimized backbone structures at high resolution from an initial backbone which can be partially or entirely artificially constructed. During the optimization, generic instead of specific sidechain types can be employed, solving the problem of designing backbones without knowing the amino acid sequence in advance.
SCUBA-D
SCUBA-D is a method based on deep learning for generating protein structure backbone.
Pythia
Pythia is a tool for self-supervised prediction of protein mutational effects.
EnzyPick
Enzyme screening is an essential preliminary activity in metabolic engineering. Nonetheless, the existing tools largely rely on prior knowledge, and cannot utilize custom candidate enzyme libraries. To address this, we introduced the Substrate–product Pair-based Enzyme Promiscuity Prediction (SPEPP) model, which leverages transfer learning and Transformer architecture to illuminate the intricate interplay between enzymes and substrate–product pairs. SPEPP exhibited good predictive ability, eliminating the need for prior knowledge of reactions and allowing users to define their candidate enzyme libraries. Owing to its adaptability, SPEPP can be seamlessly integrated into various metabolic engineering applications including, but not limited to, substrate/product screening, de novo pathway design, and hazardous material degradation. To better assist metabolic engineers in designing and refining biochemical pathways, particularly those without programming skills, we designed EnzyPick, an easy-to-use web server for enzyme screening, based on SPEPP.
RxnFinder
Microbial cell factories have a lot of important and promising applications in producing bulk chemicals, natural products, biofuels, and so on. The new bottleneck of microbial cell factory is how to design reaction, enzyme, and pathway, based on enormous biosynthesis data.Our team has been focused on the construction of data-driven biosynthesis design platform ( http://www.rxnfinder.org), which is composed of the following sections: (1) Biochemical reaction database (RxnFinder): our team manually curated more than 300,000 biochemical reactions, which are 10 times larger than the KEGG reactions, from more than 550,000 biosynthesis references retrieved from PubMed using more than 40 biosynthesis related keywords. More than 10 third party databases are linked, and more than 10 informatics methods are provided. (2) Enzyme discovery (ECAssignment): a chemical transformation-based method (ECAssigner) is proposed for enzyme discovery using biochemical reaction difference fingerprints and reaction similarity.(3) Biosynthetic pathway design (BioSynther): BioSynther tool was developed to design biosynthesis pathways between starting molecules and target molecules, in which users can interactively re-design biosynthetic pathways. One of the most promising applications of biosynthetic methods is to produce chemical products of high value from the ready-made chemicals. BioSynther is also developed to explore the biosynthetic potentials of precursor chemicals using BKM-react, Rhea, and more than 300,000 in house RxnFinder reactions manually curated. (4) Cell -based pathway optimization platform (SynBioEcoli, EcoSynther, LifeSynther): Based on the comprehensive biosynthetic data curated above, whole-cell modelling methods are proposed to optimize heterogeneous biosynthetic pathways. The proposed data-driven one-stop informatics platform could be used as a useful tool in metabolic engineering, biosynthesis, and synthetic biology.
Measurement methods
Acknowledgement
National Program on Key Basic Research Project (973 Program)
The National Key Research and Development Program of China
International Partnership Program of Chinese Academy of Sciences
Biological Resources Programme, Chinese Academy of Sciences